
Regenerative medicine requires a high degree of informed control in order to ensure that the critical quality attributes of the products are maintained batch after batch. Official guidance in the United States and Europe must be combined with a current working knowledge of best practice. Such knowledge will enable innovators to build production systems with confidence. This chapter briefly examines the implications of Good Manufacturing Practice legislation and guidance and the role of Quality Assurance in the generation of safe and effective cell-based therapeutics.
1. The origin of Quality Assurance (QA) and Good Manufacturing Practices (GMP)
The complexity of modern medicines and their potential to do harm if made inappropriately makes the need for assurance of quality appear obvious. This was not always the case. Improvements in the safety and quality of medicines have nearly always been made as a corrective action to tragic events [Patel 2008]. The three key characteristics of a good medicinal treatment are quality, safety and efficacy (QSE). The absence of these characteristics is not necessarily apparent to the user or to the prescribing doctor. Both place their faith in a system of controls extending from procurement through development and production to the management of transit and delivery.
For reasons of brevity this chapter focuses on QA and GMP in two of the territories comprising the International Conference on Harmonization (ICH): the United States and the European Union.
QA is sometimes confused with Quality Control (QC). QA refers to the system of management that enables an organisation to comply with the required quality standards (imposed or self-set). The collected methods for management of QA are referred to as the Quality Management System (QMS). QC is the activity required to measure the compliance of goods with the agreed specification.
GMP is the set of measures applied under the QMS to ensure QSE for medicinal product manufacture. The nature of GMP is defined by regulators in each territory and the definitions comprise a minimum set of practices to ensure compliance.
A distinction may be made between the acronyms GMP and cGMP. Although the terms may loosely be used interchangeably, cGMP is the acronym used by the American regulatory authority, the Food and Drug Administration (FDA), to refer to ‘current Good Manufacturing Practice(s)’. ‘Current’ makes explicit the expectation that developments may be made over time to improve control and so this acronym will be used here irrespective of territory. cGMP refers to controls over the processes of manufacture and the resources applied. Physical facilities are the most obvious resource but the human resources and process documents (instruction and records) must be properly controlled as well. Management of these resources and scrutiny of the effectiveness of controls require separation in the management structure between the QA and Production teams.
The reader should bear in mind that the evolution of cGMP depends on the history of standards and regulation supplemented with learning from real-life incidents. This means that some issues will be poorly defined and book learning must be supported with knowledge from custom and practice. Good QA relies on current knowledge and the support of a good professional network through which to learn best practice.
Many of the cGMPs for cell-based therapeutics (CBTs) are based upon practice for pharmaceuticals. US and EU readers are directed in the first instance to the ICH cGMP guidance [ICH Q7A].
2. Proof of training and competency
Human resources form the backbone of cGMP. International guidance [ICH Q7A] emphasises operator training. This must be appropriate to the operator role and with enough background to cGMP to place the work in context. Refresher training must be frequent enough for competency to be maintained. Training must be conducted by suitably qualified professionals. In the US the requirement is prescribed in the Title 21 of the Code of Federal Regulations (CFR)[FDA Part 211]. In the EU it is stipulated in Article 7 of the cGMP Directive [EC 2003/94]. Training is best conducted on the basis of risk assessment. Certain areas, such as aseptic production (see Stem Book chapter on Cleanrooms [Chandra]), warrant a fail-safe scheme i.e. scheduled dis-qualification of staff from operational duty. It can be helpful to use a ‘traffic light’ training control system in which an operator's qualification status is established at ‘green’ when they have been adequately trained to operate under minimal supervision. After a prescribed interval the status of the operator's training passes to ‘amber’ and they are no longer entitled to operate without direct supervision. The operator will receive an automated notification to enlist for refresher training before the date when their status passes to ‘red’ and they will no longer be permitted to do such work until re-trained.
A calendar-based re-training scheme poses the risk of inadequate training for operators who may be absent due to illness, maternity leave etc. Such a catch-all approach may result in unnecessary training for staff members who have little interaction with the subject of the training. A better approach is to use a ‘training matrix’ based upon job roles and levels of risk and that shows the extent to which each role requires training in specific aspects of cGMP. The matrix can be used as part of the change control process (see Section 3). Upon either a) expiry of a refresher interval, b) appointment to a new role or c) scheduled review of performance the training status of the employee can be compared with the matrix and a decision can be made about immediate training needs. Such a comparison is best done in consultation between the Quality Unit (see Section 3), the Production Manager and the Human Resources manager.
Training Records are required. cGMP requires proof of competency together with periodic competency assessment [ICH Q7A]. Such proof can be provided in the form of examination results or by setting practical tasks. A series of such tasks from which a selection can be made is a useful inclusion in appendices to relevant Standard Operating Procedures (SOPs).
3. Site accreditation
The authority to ‘release goods to the market’ (either for clinical trials or sale) rests on two licenses. One applies to the facility; the other is specific to the product.
The authority that recognises cGMP compliance and confers accreditation in the US is the FDA and in the EU is the member state Competent Authority. Site accreditation is almost always preceded by inspection (see below). A robust QMS forms the bedrock of cGMP and is usually the first subject of an inspection. A good QMS will prescribe levels of authority in the organisation, will clarify role expectations through job descriptions and objectives related to improvement of quality and will emphasise ‘change control’. Change control mechanisms will be exemplified in the use of a well-managed document control system, usually under the management of the ‘Quality Unit’ [ICH Q7A]. Paperwork may be divided into two categories. ‘Documents’ or ‘Instructions’ tend to be prescriptive, defining roles, methods, policies, job descriptions and specifications. ‘Records’ comprise the results of activities e.g. batch records, decision records, notes-to-file and audit reports. The purpose of the document control system is to ensure that the document set is current, complete, internally consistent and protected from unauthorised tampering. To this end there must be clarity about the version number, current copy and basis of authorisation of Documents and Instructions. The most prominent parts of the document control system are the SOPs, the validation history, calibration records, batch records and the reports of QC.
Alterations to Documents result in alterations to established practice and will follow a cycle of ‘plan’-‘execute’-‘review’-‘implement’ (the last point meaning to implement further changes where necessary. A Document Review process should be established in which Documents are formally issued by the Quality Unit and appear on a list for review at specified intervals (a two year interval is often applied). Changes to Documents should be carried out by the person best placed to understand the implications of the change. The revised Document should be reviewed by the Quality Unit for consistency in relation to other procedures. The issue and re-issue of Documents must be managed centrally. The copy to be regarded as the official, issued copy should be specified in the Policy or SOP for Document Control. Copies of superseded Documents must be retained in order to interpret past events and Records. Consideration must be made of the impact of damage to the collection and a ‘Disaster Recovery’ system should be set up in order to re-instate the system in the event of fire, flood, malicious damage etc.
Unsurprisingly the design of the cGMP cleanroom lies at the heart of the capability. The salient design features are summarised in the accompanying StemBook chapter “Cleanroom Environments design for Autologous Therapy” [Chandra].
Creation of a QA/cGMP system should ideally proceed in co-ordination with site construction. The establishment of a QA/cGMP system for a production facility for CBTs may be regarded as proceeding in three stages. The generic events under each of these headings are described in Table 1. Specific detail and order of some events may vary from one facility to another.
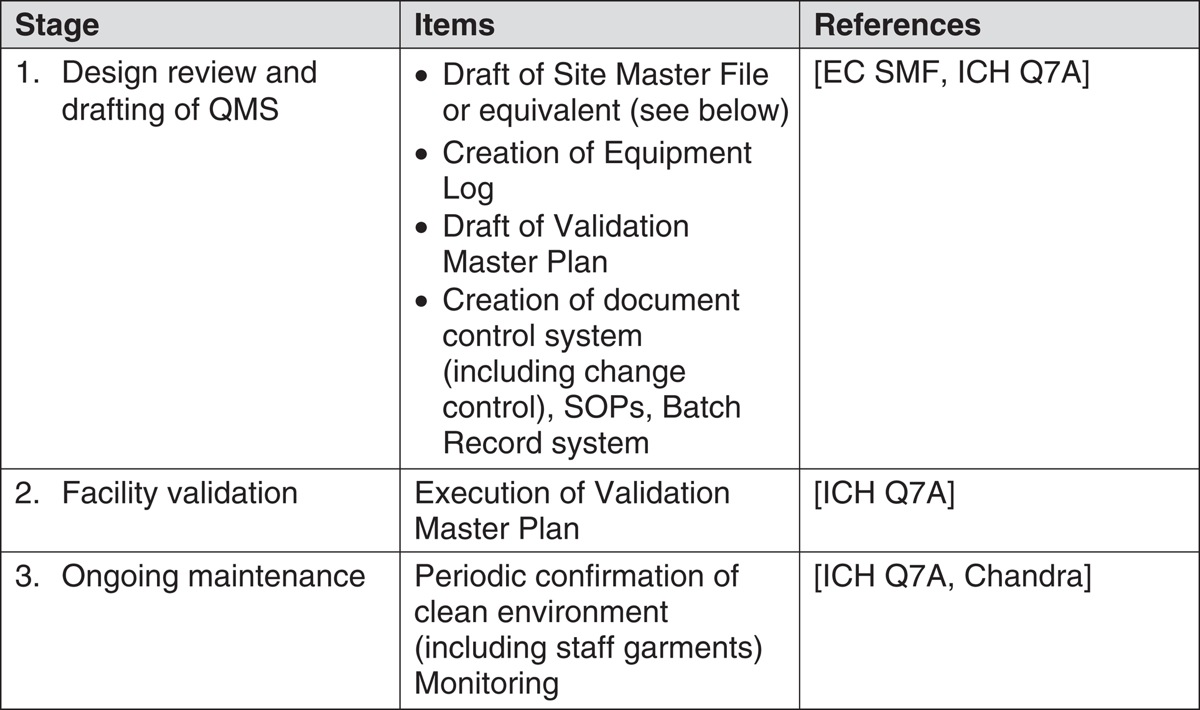
Typical stages in establishment of a GMP facility
It is good policy to record the salient features and management organisation in a master document for communicating with any inspection authority. While such a document is voluntary in both the US and the EU in practice it is expected. The ‘Site Master File’ (SMF) is an EU requirement with parallels in the US practice of issuing a ‘Type V Drug Master File (DMF)’. These are relatively short documents (typically no longer than 25 pages) and act as the primary reference for the policies, procedures and activity at the site described. The SMF is used as a reference by inspectors when planning inspections and is site-specific. It is subject to review and update. An important component of the SMF is the description of the management structure and the roles and responsibilities of members of staff. The SMF also states the policy on all important aspects of operation including
-
control strategy for processes
-
roles and relationships of the persons responsible for releasing goods from quarantine and onto the market,
-
role and activities of the QC department,
-
arrangements for contract services,
-
policy for management of complaints and recalls,
-
management of documents and records,
-
policy for self-inspection
The SMF will contain a section describing the supply-chain arrangements for the site with particular reference to the controls to avoid risks from transmissible spongiform encephalopathies and counterfeiting of raw materials and starting materials. Possibly the most detailed section of the SMF contains the validation policy of the organisation. Here the equipment is listed and the Validation Master Plan (VMP) is described. The VMP is the prescriptive statement of how the various elements of the production system, environmental and equipment, are to be validated.
A US manufacturing site carries certain obligations with respect to the FDA. This also applies to non-US sites from which goods will be imported into the US. An establishment that manufactures CBTs that qualify as drugs (most CBTs will do so), including cells from the haematopoietic or reproductive systems, is manufacturing a product subject to Section 351 of the Public Health Service (PHS) Act and is required to operate to cGMP. Compliance with the prescribed US regulations is mandatory and forms the basis of possible inspection [CFR 210, CFR 211]. 21 CFR 210 prescribes the general cGMPs and 21 CFR 211 prescribes those for the finished products. Those centres that manufacture products subject to Section 361 of the PHS Act must register and list the products so produced [CFR 1271] in order to comply with Good Tissue Practice (GTP). GTP applies to such issues as the establishment of donor eligibility and the features of subsequent processing of cells and tissues that may influence the transmission of communicable disease. This ‘Establishment Registration’ process applies to both manufacturers of blood-derived products [FDA 2830] and to manufacturers processing tissue [FDA 3356]. The distinction between ‘Section 351’ and ‘Section 361’ products above is based on risk assessment. ‘351’ products are, broadly speaking, biological medicinal products whereas ‘361’ products typically comprise transplantable tissue or blood derivatives that have been manipulated only minimally.
In situations where ‘351’ products are to be made there is no requirement for site licensing by the FDA prior to launch. Manufacture of goods for clinical trial purposes will take place under an Investigational New Drug authorisation and there may be no inspection if the FDA is satisfied by the application and supporting evidence in the ‘Chemicals, Manufacturing and Controls’ section of the IND Application, where details of the facility and process are included. In order to manufacture goods for sale, however, a full ‘Biologics License’ is required and this is attained following satisfactory completion of clinical trials, submission of a biologics license application and thorough review by the FDA.
In the EU the accreditation takes the form of a Manufacturing Authorisation and a Marketing Authorisation (the latter is often referred to as a Product License). The Marketing Authorisation is issued in response to a successful submission of a ‘Common Technical Document’ (CTD); a document that contains information about the manufacturing process and the facility and equipment. Inspection of a site for compliance with cGMP, if successful, results in the issue of a ‘GMP Certificate’ by the Competent Authority. Such certificates apply just to the site and are issued in the context of the product types that are made there. The certificates, or a record of non-compliance if appropriate, are entered into a central database for transparency [EMA].
QA management has a central role in assuring continued compliance to the prescribed standards. The mechanisms for doing this can be summarised under these headings:
-
Regular review of controls and history of the performance of the accredited areas through maintenance of an internal inspection process
-
Coordination of regular Management Review of trends in site compliance, manufacturing controls and emerging changes in standards
-
Hosting inspections
-
Oversight of the Document Control system
-
Response to Deviations and oversight of Corrective and Preventative Actions (see below)
-
Arrangement for product recalls, if required
-
Management of QA aspects of dialogue with the accrediting bodies
4. Applicable Quality Management Systems
Although the intentions for QA and cGMP are broadly the same across the ICH region there are differences in the way that the applicable regulations, standards and guidance has been written and this extends to differences in control of goods entering the market. Table 2 shows a comparison of the main features of control for the US and EU regions.
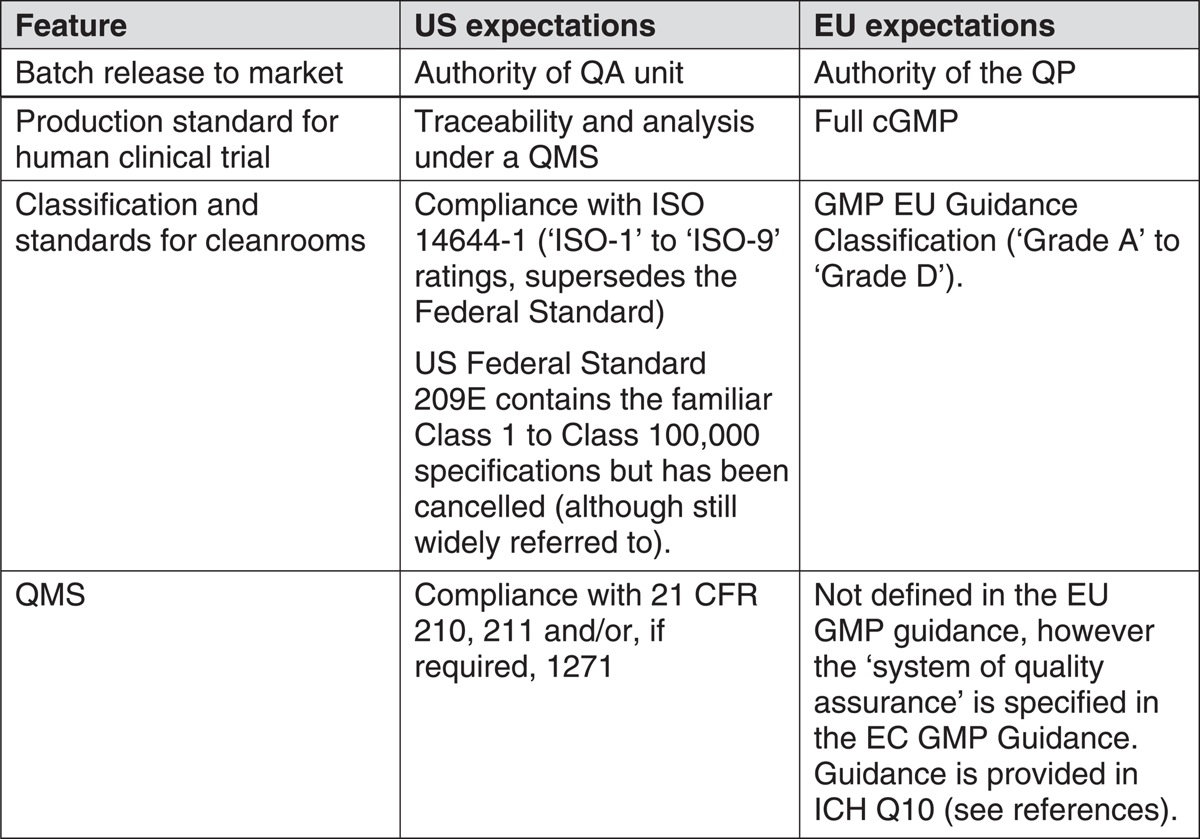
A comparison of the salient points in QA and cGMP regulation in the US and the EU
For a site embarking for the first time upon the development and launch of a CBT it may be pragmatic to develop the QMS in alignment with the phases of clinical development. For an established site with a steady flow of products through the development pipeline a decision must be made about what levels of control to invoke at what stages of development. Some businesses develop in-house levels of QMS that increase in stringency as development stage-gates are passed. This requires careful design and good operator discipline, sometimes segregating operators at different stages of QMS by department, in order to avoid inadequate control at later stages of development.
In the US in addition to the cGMP accreditation it may be desirable to gain accreditation to the standards issued by the American Association of Tissue Banks [AATB]. This is a voluntary scheme but will provide a high level of confidence in control of each aspect of tissue procurement, processing and storage. In the EU requirements vary. For example the UK requires that compliance with the Human Tissue Act be observed for research and development using primary cell isolates.
Any QMS for CBTs should have the joint objectives of maintaining QSE and ensuring that there is no transmission of communicable disease. The QMS may be paper-based or electronic but, whichever format is used, the primary records must be managed so as to protect them from post hoc tampering. If electronic records are used a key feature must be compliance with the electronic signatures regulations [CFR 11] in order to show unique traceability to identifiable, responsible individuals.
The security provided by 21 CFR 11 was seen by some researchers as being a potential impediment to the application of new technology in process development. The revised guidance [CFR 11] made it clear that legacy systems (i.e. those operational and appropriately documented before 20th August 1997) would not be subject to compliance inspections. The guidance also reinforced the requirement that careful documentation be made of the decision to implement either paper or electronic records as the primary quality record for the purposes of the data trail.
A common question for organisations embarking on cGMP process development is “How can I obtain GMP-grade raw materials?” Put simply it is the responsibility of the developer to procure raw materials with appropriate investigation and control of the quality aspects, including safety aspects related to transmissible diseases. For this reason it is prudent to apply Quality Agreements to upstream suppliers of those raw materials and consumables that are critical to quality. Without such agreements the possibility exists that the supplier may make changes to the quality of supplies which may move production controls outside the Design Space (described below), particularly if the supplier is unable to appreciate the reason why such a change may be significant to the customer.
The Foundation for the Accreditation of Cellular Therapy (FACT) has, with the Joint Accreditation Committee-ISCT, Europe (JACIE) produced a set of standards aimed at ensuring quality in medical and laboratory practice for stem cell transplantation [FACT-JACIE]. The Standard sets out minimal requirements and may be enhanced for specific centres. It covers all activities from collection to administration, whether minimally or more-than-minimally manipulated cells are used. JACIE conducts inspections and issues certificates of compliance to recognise good practice. Compliance with the Standard is voluntary and enables an operation to demonstrate that it has an excellent and appropriate QMS. The Standard is aligned with both US and EU requirements subject to the usual caveats concerning current practice. The European Commission has recognised and supported the work of JACIE and, in 2004, undertook a programme to harmonise the Standard with the content of Directive 2004/23/EC.
5. Management structure
The ICH guidance on cGMP [ICH Q7A] states that “The responsibilities of all personnel engaged in the manufacture of intermediates and APIs should be specified in writing”. A general principle to be observed is that staff members who are directly responsible for production duties should have a reporting line to senior management separate from that for the Quality Unit (i.e. the QA and QC staff). This is to avoid conflicts of interest in the day-to-day management of production especially with respect to decisions regarding batch release.
FACT-JACIE [FACT-JACIE] and the International Society for Cellular Therapy [ISCT] have published a Standard and template Quality Plans respectively that offer useful guidance on reporting lines and personnel structures for organisations involved in collection, processing and delivery of CBTs.
In the case of the EU the special risks and responsibilities associated with release of goods to the market is recognised by the requirement for a designated ‘Qualified Person’ (QP). The QP is employed by the business but is effectively accountable to the Competent Authority in the Member State. The requirement for a QP, who must be named on the Manufacturing Authorisation, is made in the Medicines Directive [EP 2001/83/EC]. The requirement for product release by the QP has been extended to goods for use in clinical trials [EP 2001/20/EC].
6. Inspections and audits
An organisation operating to cGMP will carry out a schedule of self-inspections as a part of the quality management activity. In addition the organisation will be subject to inspection from regional authorities. There are three types of such inspections:
-
Pre-approval inspections (see ‘Site Accreditation’ above)
-
Periodic, comprehensive cGMP inspections (typically biennial)
-
‘For cause’ inspections
In the US the principle of full biennial inspections as a minimum was established in the Food, Drug and Cosmetic Act. In practice a pragmatic approach has been operated since 2005 in which a risk assessment is used to assign priority for inspection to manufacturing establishments. This allows for ‘abbreviated’ and ‘full’ inspections. Inspection concludes with the issue of Form FDA 483: ‘Inspectional Observations’. The Form is discussed with management as it contains details of the degree of current compliance classified as ‘No Action Indicated’ (NAI), ‘Voluntary Action Indicated’ (VAI) and ‘Official Action Indicated’ (OAI). An OAI is usually accompanied by a Warning Letter to which the organisation has 15 days to respond. If the deficiencies are not addressed the FDA may respond without further warning, imposing criminal penalties.
The FDA operates a systems-based inspection approach structured around:
-
The Quality System
-
The five ‘manufacturing systems’:
-
Production
-
Facilities and Equipment
-
Laboratory Control
-
Materials and Packaging
-
Labelling
-
This system aligns with the structure of the subchapters of the cGMP regulation in the US.
At the international level co-operation in standards of inspection is encouraged through two mechanisms: the Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme (PIC/S). The Convention had its origin in the European Free Trade Association initiative “The Convention for the Mutual Recognition of Inspections in Respect of the Manufacture of Pharmaceutical Products”. The Pharmaceutical Inspection Co-operation Scheme was formed in 1995 in response to the arrival of new Accession States, allowing for the lack of alignment between European Law and the Convention. The Pharmaceutical Inspection Co-operation Scheme is an informal arrangement between national health authorities. Publications by PIC/S form a valuable source of reference for Quality Assurance professionals.
7. Corrective And Preventative Actions (CAPA)
CAPA is that part of the QMS that manages the response to errors, accidents, complaints and ‘adverse events’. The response must be robust, appropriate and timely and is one of the most important elements in an effective QMS. Guidance on CAPA is laid out in ICH Q10. FDA reporting guidelines are given in 21 CFR 1271.350. Instances of formal application of the CAPA process should be reviewed at regular Management Review meetings and assessed for the appearance of trends that may justify continual improvement measures. See also section 10 concerning recall.
8. Storage and its control
Storage must be arranged for the following categories of materials.
-
Packaging materials (including label stock)
-
Raw materials
-
Intermediates
-
Retained samples
-
Finished product
Sufficient capacity should be allowed and in the case of CBTs it is prudent to provide back-up capacity in case of equipment failure for materials that require low temperature storage. Depending on the value and status of the inventory it is good practice to establish an alarm system with warning levels within the range of critical temperatures. A call-out rota of qualified members of staff is an essential part of this provision. CBT manufacture generally requires a perishable raw materials inventory and low temperature finished goods store and so it is important to provide a back-up power supply, preferably with an uninterruptible power supply to bridge the momentary pause between power failure and start of back-up generator to ensure continuity of digital monitoring and control. Storage areas should be subject to regular performance review and inspection, including assessment of potential ingress of weather or pests.
The status of the goods in inventory should be clearly signed e.g. products in quarantine, released goods, rejected goods and returned or recalled product. In particular defective goods must be stored away from those that may qualify for use. Goods in quarantine, e.g. goods inwards awaiting clearance for use or product awaiting instruction to release, should be protected from unauthorised access by measures such as locking cabinets or cages.
Records must be kept of the performance of environmental controls for the storage areas of sensitive stock, typically this comprises temperature and humidity controls. Repeated exposure of cells to ambient temperature, even without thawing, can affect cell health over time. An organisation will do well to arrange liquid nitrogen storage vessels in such a way that straws of cells are withdrawn in the order that they are expected to be used. This avoids repeated withdrawal and replacement of unused cells.
9. Document control and audit trails
The management system for Document Control must, according to the European Guidance “establish, control, monitor and record all activities which directly or indirectly impact on all aspects of the quality” of the products [EC Eudralex Vol 4]. The QMS must be detailed and prescriptive enough to specify how the documents are maintained, how the current versions are issued, reviewed, updated and withdrawn and to define the levels of authority required to manage aspects of the system. The types of documents must be defined and these definitions must be adhered to in practice.
Instructional documents must carry the date from which they are effective and must be issued by identifiable persons with an adequate level of authority.
Records (see above) must be completed at the time of the events to which they refer, must be clearly legible and must be traceable without ambiguity to the person making the record. Handwritten records must be completed in indelible ink. Any additions, corrections or alterations to handwritten records must be signed and dated and the reason for the change entered on the record where appropriate. Minimum retention periods for records vary according to type of record, context and type of product, and reference should be made to applicable legislation and best practice. Many organisations establish longer-term retention policies than those required by law. It is good practice to maintain such quality records in a protected environment and to establish a disaster-recovery system. The guidance requires protection of the integrity of the record and so access to any repository should be by authorised personnel only and a record should be kept of instances of access.
Inspection of the effectiveness of the Document Control system should be part of the internal audit and inspection process. The completeness and integrity of the system should be assessed through both top-down and bottom-up sampling to ensure that there are no omissions and that the review cycles are effective.
10. Release of goods and recall
In the US the decision on batch release is the responsibility of the Quality Unit. In the EU the results of QC and the batch records must first be authorised by the Qualified Person as noted above. In either case the decision to release is based upon evidence that the product meets specification and that the Critical Quality Attributes (CQAs) of the product have been conserved. Satisfactory results from analysis to specification are a necessary but insufficient condition for product release and the example of sterility testing of a batch of parenterally-delivered cells will serve as an example of why this is the case.
Consider an aseptic filling operation in which sterile ampoules are charged with cells in buffer. Current requirements are for such a high-risk operation to be conducted in a clean environment under continuous air monitoring. A major risk for product safety is the entrainment of viable micro-organisms in one or more of the vials. If the batch (perhaps of thousands of vials) is tested for sterility by analysing a sample of the filled vials and no viable organisms are found then the batch has met specification. However, the mode of introduction of micro-organisms during filling is unlikely to be an event spread evenly across the whole operation. It will, rather, result from an intrusion, for example an operator leaning into the sterile field to adjust equipment. While shedding of viable micro-organisms may appear in the air monitoring records it is unlikely that the few vials so affected will appear in the sample sent for analysis. In fact no sample size short of 100% testing will satisfy such a requirement. The only way that the deficiency will be detected reliably is from observation of the operation and inspection of records of the environmental controls. Assurance of quality comes from robust training, robust processes and robust controls. These attributes can only be confirmed through audit, planning, inspection of records and direct observation. Hence the activity of product release is an ongoing activity in which continuity of knowledge is essential.
The concept of process robustness requires further elaboration. The quality criteria for cGMP when applied to stem cell products are derived largely from the criteria for pharmaceuticals. The development of a robust process is challenging. Consider the increase in sophistication when progressing from medical products such as devices, where the mode of action is usually apparent from the physical characteristics of the product, through to synthetic small-molecule pharmaceuticals, where the link must be made to some feature within the whole-body context and a hypothetical mode of action (MoA) is derived and proved. Once the link between MoA and structure has been made with confidence the task of confirming the necessary CQAs of the product is straightforward. The dependence of the CQAs upon process features can then be mapped and the Critical Control Points (CCPs) identified. This has two significant consequences. The first consequence is that a ‘Control Strategy’ can be determined; i.e. the combination of process controls and QC tests that provides an acceptably high level of confidence that the CQAs will be conserved. A part of the Control Strategy is the identification of a ‘Design Space’ i.e. the set of limits to the process parameters contained in the Control Strategy that provide assurance of quality. The task of process validation then consists of confirming the validity of the choice of Design Space in the production plant of choice. The second consequence is that, in the case of small-molecule pharmaceuticals, the CQAs may be attainable using more than one synthetic route. Hence a business may opt to change to a cheaper production method in the future as long as the product possesses the same CQAs and the resultant impurities or co-products are contained within acceptable toxicological limits. The biggest challenges usually arise from control of, say, crystal polymorphism or from stereoisomerism. By contrast biopharmaceuticals, including tissue- and cell-based therapeutics, are complex mixtures. It is often difficult to say with certainty what feature, or combination of features, is responsible for the activity observed during development and for the performance recorded during clinical trials. In regulatory parlance it is not possible to assert that ‘comparability’ has been maintained.
Historically there have been two methods of dealing with this difficulty; both are described in ICH guidance [ICH Q8]. The first method (referred to as the ‘minimal approach’) consists of establishing a relatively narrow Design Space, i.e. one that is restricted to the scope of variation encountered with the production equipment used for the efficacy and safety studies, and accepting the commercial risk that, if future changes become necessary that will take the process outside that design space then the business must bear the cost in money and time of conducting the additional studies necessary to establish comparability. The results will be subject to regulatory review before approval can be granted to continue with manufacture. The second method (the ‘enhanced approach’ or ‘Quality by Design’) consists of conducting wide-ranging studies that will result in enhanced understanding of the dependence of the CQAs upon process parameters inside a wider design space. Under these circumstances it will be possible to reduce the number of requests for post-approval review or process changes to a minimum and to reduce QC testing as a result of confidence in the process itself.
On 21st August 2002 the FDA launched a two-year initiative on pharmaceutical GMPs [CDER GMPs]. Amongst the objectives was the need to “encourage the early adoption of new technological advances” and to “facilitate industry application of modern quality management techniques, including implementation of quality systems approaches, to all aspects of pharmaceutical production and quality assurance”.
The PAT initiative applies to the ‘Chemistry, Manufacturing and Controls’ (CMC) part of CBT production i.e. the Control Strategy and process capability enhanced with the aid of new technologies. Organisations are encouraged to work on a voluntary basis with the FDA to avoid a ‘regulatory impasse’ that would otherwise be created by attempting to introduce unproven approaches to control. An emphasis is placed on a mechanistic understanding of the relationship between process and product specifications and the product performance. In particular the usefulness of real-time QA measures is encouraged. This is especially relevant to CBTs because of the need to avoid unnecessary delay in product release.
Product recalls may occur when a deviation or error is uncovered after release, sometimes following complaints. In the EU the basis for guidelines and controls is contained in the Directives [EP 2001/83/EC, EC 2003/94/EC]. The response to these events should be managed swiftly under a pre-established written protocol and using a risk-based decision process. Records must be kept diligently and all complaints must be recorded. The staff dealing with the complaint and/or recall must be well trained and independent of the sales and marketing function of the organisation. The QP must be kept informed and, preferably, involved in the process. Wherever possible a sample of the defective material must be retrieved from the complainant and reference should be made to retained samples when carrying out the investigation. Naturally it is important to access distribution information in order to recall all affected product if necessary. The investigation must identify the root cause of the problem and avoid addressing just the immediate consequences. Whatever the outcome of the investigation it is important to place the event in the context of previous incidents in order to decide whether more fundamental corrective actions should be taken. Operations should bear in mind that the accumulation of minor deficiencies with respect to cGMP may result, on inspection by the authorities, in notice of a major deficiency due to lack of management control.
Care must be taken to protect public safety and, if the incident is serious and may take some time to evaluate, a recall may be appropriate even before the investigation is complete. The investigation must be carried out in a timely manner and, if the result is a recall, the holder of the relevant Authorisations and the Competent Authority must be informed. Further information is available from the Eudralex website [EC Eudralex Vol 4].
11. Final thoughts
For commercial and clinical success there must be an emphasis on the economy and elegance of solutions to the requirements and to the spirit of GMP. Both over-commitment of resources and ignorance or unjustifiable short cuts must be avoided. Successful application of GMP requires both an understanding of the requirements in law and of the industry norms. Such an understanding is best obtained through long immersion in best practice and up-to-date information from the regulatory authorities and established practitioners. Many companies find it helpful to begin operations with the help of a consultant and gradually to make the transition towards full internalisation of controls.
References
Last revised June 11, 2014. Published June 30, 2014. This chapter should be cited as: Medcalf, N., Hourd, P., Chandra, A. and Williams, D. J., Quality assurance and GMP in the manufacture of cell-based therapeutics (June 30, 2014), StemBook, ed. The Stem Cell Research Community, StemBook, doi/10.3824/stembook.1.99.1.