
While many clinician- and industry-led autologous cellular therapies are demonstrating benefits to patients in clinical trials, few products have been commercially approved. Progress towards production and commercialization still faces substantial translational challenges under existing regulatory frameworks. Manufacturing and supply of more-than-minimally manipulated (MTMM) autologous cell based therapies presents a number of unique challenges driven by complex supply logistics and the need to scale-out production to multiple manufacturing sites or potentially near to the patient within hospital settings. The existing regulatory structure in Europe and the U.S imposes a requirement to establish and maintain comparability. Under a single market authorisation this is likely to become an insurmountable burden for the roll-out of manufacturing processes to more than two or three sites unless new enabling manufacturing and regulatory science can be established to bridge the comparability challenge.
1. Introduction
Cell based therapies fall into two broad classes, those derived from a patient's own cells (autologous or ‘one to one’ therapies) and those derived from a donor's cells (allogeneic or ‘one to many’ therapies). This distinction drives the product safety and efficacy model and the approaches to manufacturing, transportation and clinical delivery of the product. In turn, this dictates the technical and regulatory challenges for developing and commercialising safe, effective and reproducible cell based therapies at the required scale and cost.
Over the last ten years there has been a steady increase in the clinical development of autologous cell products. An increasing number of clinician- and industry-led autologous cellular therapies are demonstrating benefits to patients in pivotal or late stage clinical trials in many therapeutic areas, including cardiovascular disease, peripheral arterial disease, liver disease, diabetes, neurodegenerative disorders, bone repair, and spinal cord injuries (see Section 3 of this Chapter). However, if these products are to progress further down the development pipeline, a number of specific challenges associated with their manufacturing and supply need to be overcome to permit the scale/roll-out of autologous treatments under existing regulatory frameworks. This review focusses on how the existing regulations impact the development and commercialization of autologous cell therapy products. Particular emphasis is placed on the specific challenges for the manufacture and scale-out of more-than-minimally manipulated (MTMM) autologous cell based therapies to multiple manufacturing sites or potentially near to the patient within hospital settings.
2. Manufacturing and supply approaches
Manufacturing and supply of autologous cell based therapies presents a number of specific and additional challenges. As distinct from allogeneic therapy or traditional pharmaceuticals/biologics production, this is driven by complex supply logistics and the need to scale-out (replicating the manufacturing line or unit operation to increase the number of batches) rather than scale-up production (increasing manufacturing output by increasing the volume or number of cells processed for each batch).
Most companies seeking highly profitable business models work predominantly with scalable allogeneic therapies following a business and supply model similar to that of the conventional biopharmaceuticals (Williams et al, 2012; Foley & Whitaker, 2012). Smaller scale autologous therapies need to follow alternative manufacturing and distribution approaches, dependent on the product (disease indication and prevalence), the method of preservation of the product and the fit with the systems in place at the final destination in the clinic (Mason & Dunhill, 2009; McKernan et al, 2010; McCall & Williams, 2012).
Approaches may involve a central processing facility serving a number of clinical sites and which require patients to travel to a specialised centre for treatment (e.g. Tigenix have used this approach for ChondroCelect®). They may involve a distributed model that requires localised processing within a hospital unit or manufacturing in-theatre or at the bedside, in which cells are removed from patients and processed locally by means of a closed or functionally closed, automated processing system or device before being reintroduced into patient on-site (Figure 1). Allowing bedside isolation and enrichment of cells for treatment of a range of conditions, commercial examples of these near patient or point-of-care processing devices are already in use, for example, CliniMACs® Prodigy (Miltenyi Biotec), Cellution® (Cytori Therapeutics Inc.) and HiQCell (Regeneus Ltd). So far these have primarily been used for cell therapy products that are not regulated as medicinal products i.e. the cells are not substantially modified or altered and are used in a form similar to their original function (McKernan et al, 2010; Bravery, 2012).
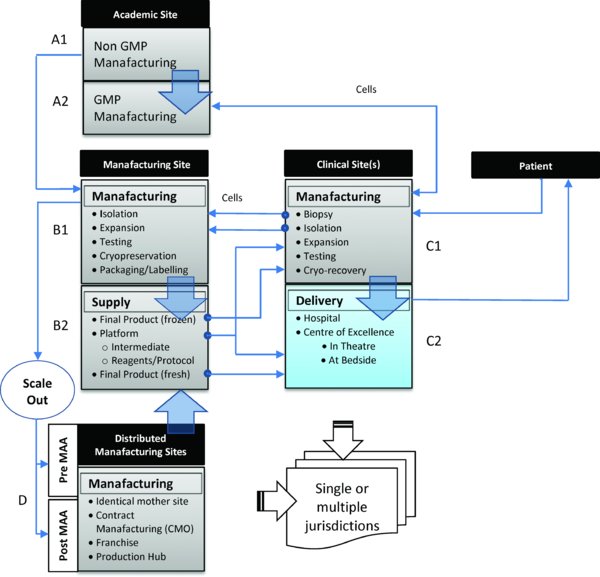
Alternative routes for manufacturing, distribution and delivery of small scale more-than-minimally manipulated autologous cell therapy products. Approaches may involve (i) a regulated central processing facility (B1 or A2) serving a number of clinical sites in which the patient travels to the clinical site/specialised centre for treatment. Cells are removed from the patient (C1) and transferred to the regulated manufacturing site (B1 or A2) before being returned back to the clinical site for administration to the patient either directly as fresh product (C2) or following further processing (cryo-recovery) (C1) or (ii) a distributed model that requires localised processing within a hospital unit (C1) or manufacturing in-theatre or at the bedside (C2), in which cells are removed from patients and processed locally by means of a closed, automated processing system(s) before being reintroduced into the patient on-site. The origin and scale of the potential routes for manufacturing scale/roll-out to multiple sites may involve transfer of a manufacturing process and product (iii) from an academic (A1) or hospital laboratory (C1) to a regulated manufacturing site (B1 or A2); (iv) to one or more additional production lines within the same facility or to a regulated manufacturing site(s) (D) within the same jurisdiction, either before or after Phase III clinical trials (i.e. pre- or post-Marketing Authorisation Application (MAA)) or to regulated manufacturing site(s) within different jurisdictions, or (v) to processing sites in International Clinical Centres of Excellence for major clinical specialisms (C2). Potential routes may also involve the roll-out of self-contained manufacturing platforms, standardised reagents and protocols (B2) close to the clinic (C2) or to local production hubs (D).
3. Clinical and industrial landscape
Research and development in cell based therapies is maturing (Fisher & Mauck, 2013) but large conventional pharmaceutical companies are seemingly reluctant to engage in ‘high risk’ early investigational phases of their development (McKernan et al, 2010). There is however some evidence that this is beginning to change (ARM Annual Report, 2013; Mason et al, 2013).
3.1. Clinical pipeline of cell therapies
In Europe, the major stakeholders developing Advanced Therapy Medicinal Products (ATMPs) are hospitals, academic institutions, charities, and Small and Medium sized Enterprises (SMEs) (Maciulaitis, 2012; HoL, 2013). Academic and clinical centres with Good Manufacturing Practice (GMP) compliant facilities are also becoming more common across Europe and the U.S. These are now significant contributors to the translation of cell therapy research to GMP compliant protocols and for the provision of non-clinical and clinical trial GMP-grade material (Maciulaitis et al, 2012; Culme-Seymour et al, 2012; Cell Therapy Catapult, 2013; Pearce et al, 2014)
The development of new ATMPs is largely investigator-led in the EU and U.S with Europe lagging behind the U.S in terms of the cellular therapies in clinical trials. Recent analysis by Foley & Whitaker (2012) has shown that an increasing number of clinician-led (i.e. clinical trials sponsored by an institution), predominantly autologous cellular therapies, are demonstrating benefits to patients. These clinician-led therapies span those where a degree of clinical adoption and proven efficacy already exists to those where trials will be carried out under regulatory constraints more familiar to company-led cellular therapies. In their study, Foley and Whitaker (2012) identified two prominent groups of the cellular therapies in development. Clinician-led autologous cellular therapies that focus primarily on procedures (defined as cell therapies with complex routes of administration) represented 63% of all clinician-led trials (437). Company-led cellular therapies that were primarily allogeneic and product-focused (defined as cell therapies where intervention is minimal) represented 44% of all company-led trials (66). Overall, of the 503 trials sampled, an estimated 87% were led by clinicians and 13% by companies. Of all the trials (involving both procedures and products), 71% were autologous (355) and of these only 22 (6%) were company-led.
According to industry data compiled by the Cell Therapy Group (Buckler, 2012), as of August 2012, there were 48 industry-sponsored (40 companies) clinical trials of cell therapies in pivotal or late stages (Phase III or PhaseII/III), of which 59% were autologous. From a U.S perspective, a recent report from the Pharmaceutical Research and Manufacturers of America (PhRMA, 2013), which lists all U.S industrial sponsored biologics in clinical trials, identified 69 cell therapies in clinical trials or under review by the U.S Food and Drug Administration (U.S FDA). Fifteen were in Phase III clinical trials, of which six were autologous. From a UK perspective, as one of the leading countries in Europe developing ATMPs alongside Germany and Spain (Maciulaitis, 2012), as of April 2013 there were 34 cell therapy clinical trials (mainly Phase I/II or II) ongoing in the UK, of which 23 were autologous (68%). The majority (76%) of clinical trials were sponsored by a research institution with only 6 sponsored by industry (Cell Therapy Catapult, 2013). These trends are consistent with a recent analysis of ATMP development in Europe (Maciulaitis, 2012).
3.2. Industrial snapshot of the autologous cell therapy field
Despite the rich pipeline of cell therapies that are in clinical development, only a limited number have been taken down the established regulatory pathway for cellular products and translated into products for market approval. Since 2009 there have only been 12 approvals; six in the U.S, one in Europe, one in Canada, one in New Zealand and three in South Korea (Buckler, 2012; ARM, 2013). Very few of these are autologous cell therapies.
Currently, MACI (matrix-induced autologous chondrocyte implantation), intended for the repair of cartilage defects (Genzyme/Sanofi, France) and ChondroCelect® (TiGenix, Belgium) are the only licenced cell-based ATMPs on the market in Europe. Licensed by the European Medicines Agency (EMA) in 2009, ChondroCelect® (TiGenix) as the first ATMP, is likely to set the standard for the clinical development of cell based therapeutics (Warren, 2013). In the U.S, three autologous cell therapies have been approved. Carticel® (autologous chondrocytes for cartilage repair) was the first approved product in 1997 (Genzyme/Sanofi). An autologous treatment for prostate cancer (Provenge®; Dendreon) was approved in 2010 and has been developed with a multiple manufacturing site model in mind. In addition, an autologous fibroblast product for filling wrinkles was approved in 2012 (laViv; Fibrocell). In South Korea, two autologous programmes have received approval in the past few years, an autologous bone marrow–derived cell therapy for myocardial infarction in 2011 (HeartiCellgram®-AMI; PharmiCell Co. Ltd) and an adipose tissue–derived cell therapy for anal fistulas in 2012 (Cupistem; Anterogen). It is worth noting that limited peer-reviewed clinical trial data for these products are publically available (Wohn, 2012; Bersenev, 2012).
4. Regulatory pathway for autologous cell therapies
The regulatory approach taken for specific autologous cell therapies is dictated by their intended clinical use, method of clinical delivery and manufacture. In some therapeutic cases, particularly in the orthopaedic and cosmetic sectors, harvested cells are minimally manipulated (e.g. by aseptic enrichment or separation techniques) and returned to the same patient. In most others there is a requirement to expand the number of harvested cells in in vitro culture to generate a sufficient dose for therapeutic use. This expansion in culture, being considered by regulators to be more than minimal (or substantial) manipulation, raises considerable hurdles and challenges for both developers and regulators (Ahrlund-Richter et al, 2009; CAT, 2010; Salmikangas & Celis, 2011; Van Wilder, 2012; Bravery, 2012).
Under the existing regulatory framework, cellular products that have been subject to more-than-minimal manipulation and/or do not carry out the same function in the recipient as the donor (non-homologous use) are broadly classified as either medicinal products (EU) or biologics (U.S). Relatively few regulatory distinctions are made between autologous and allogeneic therapies and the characteristics that differentiate them (U.S FDA, 2001; European Commission, 2007). In the EU and U.S, the use of such cell based medicinal products is regulated under Public health legislation and Pharmaceutical legislation. Other related legal requirements and guidelines are applicable across each stage of the product development process. For a summary of the EU and U.S legislation applicable to the development and use of human cells and tissues in human therapeutics, readers are directed to the Publically Available Specification (PAS 83:2012) published by the British Standards Institution (BSI, 2012).
The regulatory route is determined by criteria which differ between the EU and the U.S. In the U.S, cell-based biologics are regulated by the Center for Biologics Evaluation and Research under Title 21 of the Code of Federal Regulations (CFR) Part 1271 as Human Cells, Tissues and Cell and Tissue-based Products (HCT/Ps) and must gain approval from the FDA via the submission of a Biologics Licence Application (BLA) (U.S FDA, 2001). Once submitted, a BLA is subject to review by the FDA (6-10 months) to assess the quality, safety and efficacy of the product. The FDA is responsible for all facets of regulating cell-based therapies, including clinical trial authorisation and compliance with the current Good Manufacturing Practice (cGMP) requirements. The situation in Europe is somewhat more complex.
In Europe, cell-based medicinal products are regulated under the Advanced Therapy Medicinal Product (ATMP) Regulation (European Commission, 2007), which mandates that all ATMPs are subject to a centralised marketing authorisation procedure. All marketing authorisation applications are subject to a 210-day assessment procedure by the EMA, supported by the Committee for Advanced Therapies (CAT), before a licence can be granted. Member states still retain responsibility for authorisation of clinical trials occurring within their borders, and have the option to exempt certain products used on a non-routine basis for unmet clinical need (in particular the ‘Hospital Exemption’).
In the UK, the Medicines and Healthcare products Regulatory Agency (MHRA) is the supervisory authority for UK manufacturers or importers of centrally authorised ATMPs. The MHRA is the competent authority for ATMPs that are prepared and used under the Hospital Exemption and made and supplied under the ‘Specials’ scheme (see Box 1). It is also the competent authority for the assessment of applications for clinical trial authorisations and the associated manufacturer's licence for investigational ATMPs.

A cell therapy that is classified as an ATMP, regardless of it being investigational, must comply with GMP for medicinal products under Directive 2003/94/EC. GMP is defined by the MHRA in the UK as “that part of quality assurance which ensures that medicinal products are consistently produced and controlled to the quality standards appropriate to their intended use and as required by the marketing authorization or product specification”. Both the U.S. Food and Drug Administration (U.S FDA) and the European Medicines Agency (EMA) have similar definitions. As defined, GMP regulations and guidelines cover nearly all aspects of product manufacturing, including the quality control and assurance system, manufacturing facilities, equipment and devices used in the process, raw materials, media and medium supplements, storage, and shipping (Eaker et al, 2013).
Typically autologous cell-based therapies are designed to address rare conditions. A number of specific fast track or orphan designation regulatory pathways have emerged, intended to accelerate the delivery of cell therapies into routine clinical practice for unmet or poorly met medical conditions and ultra-rare or life-threatening diseases. These include the fast track, priority review and accelerated approval programmes in the U.S, which have recently been expanded under the FDA Safety and Innovation Act 2012 (FDASIA) to allow sponsors to request that their therapy be designated as a “Breakthrough Therapy” (Sawyers et al, 2012). The EMA has similar licensing flexibilities, including ‘conditional approval’ and ‘accelerated assessment’ procedures and an ‘exceptional circumstances’ procedure, which provides for limited clinical development in situations where comprehensive efficacy and safety data is not feasible (EMA, 2005).
Other nations, including Japan and South Korea, are tuning their regulatory infrastructures to consider different evaluation approaches based on adaptive licensing or conditional marketing approvals. These approaches are grounded on stepwise learning and iterative phases of data accrual and regulatory re-evaluation, which allows commercial sale in certain instances whilst pivotal trials are underway.
In South Korea, for example, this has recently led to the approval of an autologous bone marrow-derived mesenchymal stem cell (MSC) therapy product (HeartiCellgram®-AMI; PharmiCell Co. Ltd) and the world's first allogeneic, off-the-shelf MSC-based product (Cartistem®; Medipost Co. Ltd). According to Ancans (2012), a similar regulatory decision has seemingly been adopted for Osiris Therapeutics Inc. allogeneic MSC product; Prochymal®. In May 2012, the company was granted an authorization for the treatment of acute graft-vs-host disease (GvHD) in children under Health Canada's Notice of Compliance with conditions (NOC/c), which is an authorization to market on condition that the manufacturer undertakes additional studies to verify the clinical benefit. It should be noted that Prochymal® has since received approval by the New Zealand Regulatory Agency (Medsafe) under their priority review scheme and is currently available in the U.S under an ‘Expanded Access Programme’ (so called “compassionate use”). It has also received an Orphan Drug designation in the EU and will be evaluated in Switzerland under their agency's (Swissmedic) ‘Rapid Authorisation Procedure’. In Japan, new Regenerative Medicine law (enacted on 20th November, 2013) directs the Ministry of Health, Labour and Welfare (MHLW) to adopt procedures that permit an accelerated clinical development pathway for regenerative medicines and living cell therapies. This is expected to fast-track therapies using a process focussing primarily on the safety profile of the cells. In practical terms this means that regenerative medicine products could conceivably reach the market after a Phase II clinical trial (Mack, 2013).
5. Regulatory challenges for the manufacture of MTMM autologous cell therapies
While many clinician- and industry-led autologous cellular therapies are demonstrating benefits to patients in clinical trials, there are persistent regulatory challenges and uncertainties impeding their prospects for translation and commercialization. Specific issues relate to the regulatory cost burden and timelines involved in the transition to GMP and in establishing product comparability for the subsequent roll-out of successful autologous cell therapies to multiple sites (McCall & Williams 2012; Williams et al, 2012). The situation is compounded by uncertainty associated with the lack of harmonisation in the way regulations are implemented and interpreted across the major geographical markets.
Market approval for specific and individualised therapies under regulations that impose expensive industry standards designed principally around the requirements of more conventional pharmaceutical products is not easily achieved. Many commentators have argued the regulations do not take into account the special characteristics of these products or the practical and resource limitations of the majority of small scale manufacturers involved in producing these therapies (Maciulaitis et al, 2012; Pirnay et al 2013; HoL Report, 2013). Indeed following the recent public consultation on the ATMP regulation in Europe (European Commission, 2013), the level of regulatory requirement was blamed for the disappearance of some innovative products from the market. Contributors also considered this as preventing the majority of developments in this area from going beyond the “Hospital Exemption” (or ‘specials’ type of manufacture in UK) or other derogations under national law. This has created a fragmented market in the EU making it difficult for companies to forecast and manage associated costs and resources. This was a factor influencing Cytori's recent decision to conclude ADVANCE, its European clinical trial for acute myocardial infarction (Cytori Press Release, 2013).
5.1. Regulatory harmonisation
The lack of regulatory harmonisation has been cited as a major hurdle in the development of cell therapies (HoL Report, 2013). Although there are initiatives, including the International Conference on Harmonisation (ICH) and the formation of a joint EMA-FDA committee, aimed at harmonising regulatory standards globally, there are many examples where there is still discord.
In Europe, the implementation of the ATMP regulations is heterogeneous between member states, with inconsistencies in the way that ATMPs are defined or classified and in the way article 28 of Regulation (EC) No.1394/2007 (the so-called Hospital Exemption clause) is interpreted. There are also diverging practices in Europe, with some member states having different national laws that exceed EU directives and regulations (European Commission, 2013; Pearce et al, 2014).
Comparing the regulatory landscape in Europe and the U.S there are several disconnects in expectations between the FDA and the EMA. The requirements for GMP compliance are not always interchangeable, with subtle differences in the sterile processing, documentation and quality control requirements. Disparities exist in the control of starting materials, sterility testing and the environmental monitoring of GMP suites, in particular the terminology and specifications for allowable levels of particulates and the measurement of microbial contamination (Ginty, 2012; Medcalf et al, 2014; Chandra et al, 2014). In terms of clinical trial regulation, there are differences between the Clinical Trial Authorisation (CTA) procedure in the EU and the Investigational New Drug (IND) application in the U.S (BIS, 2012). In contrast to the EU, GMP facilities for manufacturing of Phase I/II and phase II trials in U.S are not subject to inspection. A manufacturing authorisation (MA) is required in the EU for the manufacture of investigational medicinal products (IMP). Under the MA, each batch of a medicinal product must be released by a Qualified Person who must assess release criteria and adherence to GMP regulations and guidelines. In the U.S, there is no such requirement (Pearce et al, 2014). In fact, guidance provided by the FDA for the manufacture of some drugs and biologics for Phase I trials provides a more graded approach to GMP in which manufacturers may be exempt from many of the requirements in CFR Part 211 of the regulation (U.S FDA, 2008).
5.2. Point-of-care manufacturing
One example of an emerging area that is unclear within the existing regulatory landscape is that of closed or functionally closed systems applied to the manufacture of an autologous cell therapy product at the bedside or in a hospital setting. Certain near patient or point-of-care processing devices are already in use, but these have primarily been used to enrich cells from a tissue/cell fraction before being returned to the patient within the same surgical procedure without culture and manipulation (see Section 2).
However, if the cells are MTMM and/or used for a non-homologous application, the question arises as to how they will be regulated, dependent on whether the system is to be validated as a piece of equipment or as a device for producing a product licensed under ATMP or HCT/P regulations. The challenge from a regulatory perspective will be in determining where GMP starts for a bedside device producing an ATMP or HCT/P, how these systems or machines can be inspected and how a Qualified Person (QP) can release final product for immediate administration into the patient. If U.S and EU regulatory pathways for licensing these systems can be clarified, they may offer an attractive solution to the logistical issues posed by traditional manufacturing supply models for autologous cell therapies, whether located in the hospital setting or the distributed manufacturing setting.
5.3. Multisite manufacture – the hidden challenge of comparability
In the autologous setting, logistical hurdles are associated with the clinically limited time available to transport harvested donor patient cells to the manufacturing site and their return back to the clinical site for administration to the patient. This dictates both the manufacturing model (centralised vs distributed) and the clinical-site model (direct delivery vs clinical-site manipulation), which present a number of ways of realising the manufacturing/clinical supply process in multiple distributed locations (Figure 1). The requirements for regulatory approval, GMP compliance and the level of validation relate in part to which sites are used for each element of the manufacturing and clinical process.
Building quality standards and GMP into the development process means that cell processing facilities must meet structural and design specifications making it fit for purpose as well as demonstrating a GMP compliant quality system (Medcalf et al, 2014). This can be a challenge for small developing companies and product developers in academic or hospital settings. In many cases they are bound to using contract manufacturers or are transitioning from non-GMP facilities or facilities accredited as tissue establishments for which the environmental requirements are not usually compliant with GMP. This raises difficulties in manufacturing continuity during later phases of development (Salmikangas & Celis, 2011; Baum et al, 2013) and makes the transition to commercial-scale production of autologous cell therapies, with few manufacturing economies of scale, prohibitively expensive or even unfeasible (McCall & Williams, 2012; Pearce et al 2014). The challenge that this poses is no better illustrated than by Dendreon's conspicuous and costly issues with their multiple manufacturing site model and uptake of their autologous prostate cancer product (Provenge®) (Karkaria, 2011).
The main challenge in manufacturing however is the need to scale-out production of autologous cell therapy products for both multi-centric Phase III studies and for the supply of marketed products to clinical sites in multiple distributed locations, potentially within different regulatory jurisdictions. This may involve the introduction of significant changes to manufacturing processes to replace non-GMP-grade cells or xenogeneic raw materials for example. It may involve the introduction of a second or reconfigured production line/unit at an existing site. It may involve outsourcing manufacturing via a Contract Manufacturing Organisation(s) (CMO) or franchise(s), or the construction of additional manufacturing sites. It could potentially involve the roll-out of self-contained manufacturing modules close to the clinic or to local production hubs similar to franchised operations (Figure 1).
The existing regulatory structure in Europe and the US sensibly imposes a requirement to establish and maintain comparability (a demonstration of product equivalence) between sites or when changes are made to the manufacturing process. The regulatory expectations for the degree of assurance and stringency of the data required to demonstrate comparability will depend on the nature and extent of the change and the stage of product development. This could involve extensive in vitro characterisation, with or without non-clinical or clinical testing (or both). So from a regulatory perspective, the expectation is that comparability testing programmes consider an assessment of the impact of changes to the product quality attribute profile caused by the manufacturing change, as it relates to the safety and efficacy of the product (EMA, 2005, CAT, 2010). A recent report by the Potency Working Group (formed by the ISCT Process and Product Development Committee), has asserted that without the inclusion of measures of biological activity in the testing programme (including surrogate measures of potency that allow prediction of activity in vivo), product comparability cannot be established after changes, even minor changes, are made to the manufacturing process without non-clinical or clinical testing (or both) (Bravery et al, 2013).
Even if valid and quantitative predictive surrogate tools applicable to such extensive testing were available, extensive safety testing of final cell product, while practical in the allogeneic setting, may not be feasible in the MTMM autologous setting. This is because of restrictions related to small lot sizes, short shelf-lives and the clinically limited time available for product and lot release testing, as well as persistent issues related to variability of the donor derived starting material (both intra- and inter-individual). This means that it may not be possible to demonstrate comparability for additional manufacturing sites without costly and time-consuming confirmatory clinical qualification studies.
The scale-out/transfer of manufacturing processes to multiple sites established before pivotal Phase III clinical trials may be achievable. Under a single market authorisation however, the roll-out of locked down processes to more than two or three sites is likely to become an insurmountable challenge, even within the same regulatory jurisdiction (Williams et al, 2012; Bravery, 2012). Unless alternative manufacturing approaches can be found to bridge the regulatory and scientific challenges of comparability, realising a sustainable and investable business model for affordable MTMM autologous cell therapy supply is likely to be extremely demanding, even within a clinical setting. Without comparability there can be no process change, no second process or site, no real scalability, no roll-out after success and no effective manufacturing strategy (Table 1). This threatens the progression of these products down the development pipeline and ultimately patient accessibility to an increasing number of clinician-led autologous cellular therapies currently in development.
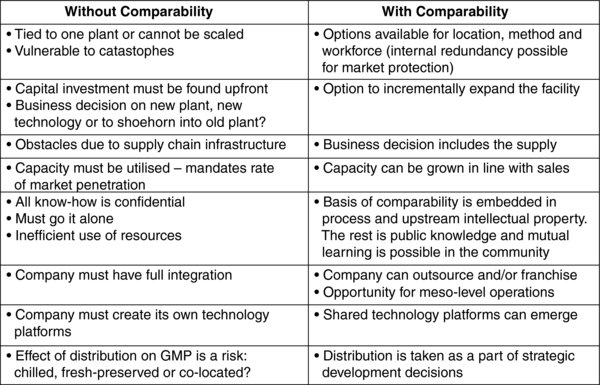
Manufacturing strategies for MTMM autologous cell based products from the perspective of process development with and without a proportionate approach to comparability.
6. Future perspectives – the need for regulatory debate
The cell therapy industry has experienced continued wrangling's between regulators, law makers and practitioners and uncertainty as to the data required to establish quality, safety and efficacy of cell therapies (Freeman & Fuerst, 2012; Lttleman, 2012; McAllister et al, 2012; Werner et al, 2012; DeFrancesco, 2012; Bianco et al, 2013; Bravery et al, 2013).
Despite the rich pipeline of autologous cell therapies in clinical development, few MTMM autologous cell therapies have made it onto the market, with no precedent for a multi-site MTMM autologous cell therapy in Europe. Given the state of the industry, it is clear that late stage development and clinical progress towards production and commercialization of autologous cell therapies still faces substantial technical and regulatory challenges.
The regulation of cell based therapeutics has matured in a manner that is an extension of the biopharmaceutical regulatory framework. The regulators acknowledge the formal complexity of these products (CAT, 2010; Maciulaitis et al, 2012; Ehmann et al, 2013; Vestergaard et al, 2013). This acknowledgement takes the form of recognition that the product is too complex for quality to be assured by testing to specification alone. Reproducibility and control of the process is important from a regulatory perspective since this has a direct impact on product performance and product reproducibility. Hence there is a special emphasis upon validation, maintenance of ‘comparability’ of production and management of any changes. This emphasis extends to those products that share some of the characteristics of Regenerative Medicine products such as the biologic/device combination products and devices for cell selection and concentration.
The current situation in Europe has led to polarisation of the approach to production of ATMPs and autologous cell therapies in particular, into two broad categories. One approach comprises the use of the Hospital Exemption (HE) clause (Article 28 of Regulation (EC) 1394/2007) which allows hospitals across Europe to produce limited quantities of such products under the exclusive authority of the medical practitioner for use with individual designated patients. At present there is inconsistency in the way the clause is interpreted. This has led to diverging practices across Europe allowing a mechanism by which locally produced ATMPs may have moved from merely exceptional or non-routine use to compete with their centrally licensed counterparts. With the potential to discourage marketing authorisation applications, this situation is viewed by some experts as a competitive disadvantage for SMEs and a threat to commercialization, as well as a potential threat to patient health outcomes (van Wilder 2012; Pirnay et al, 2013). Echoing responses to the recent public consultation on the ATMP regulation (European Commission, 2013), the HE legislation would benefit from harmonisation of the implementation of HE across Europe and a clearer definition of its scope and operational requirements in order to demarcate the commercial restrictions of the clause. There is also a need to establish how clinical sites can migrate from the occasional, as exemplified by the HE, to alternative business models focussed around clinical Centres of Excellence, especially for producing clinically pulled autologous therapies that do not have the characteristics of blockbusters or other similar commercial viability but that do lead to considerable personal and public health benefit.
The other approach is to adopt a conservative position in which manufacture is centralised according to disciplined controls on a single licensed site. The need to ensure that comparability pre- and post-launch is maintained means that development in the late stages is normally aimed at production scale. This approach imposes high capital costs upon the business at a stage where the return is uncertain. Capacity must be built before the demand will ensure full utilisation and risk to the business arises from an uncertain breakeven point for the sunk costs. In the present straitened economic climate, with poor investor confidence, this situation presents a serious barrier to entry for start-ups and SMEs.
In order to access more pragmatic models of business and production a key question arises on the extent to which the existing regulations are applicable to cell based therapies and whether all cell based therapies should be treated the same. This question is the subject of an ongoing vigorous debate and is illustrated by the perceived uncertainty within the current delineation of the regulations when defining a product as a medicine, device or combination product. Cytori's recent decision to withdraw from the European market illustrates this point. This regulatory imbalance creates specific and additional challenges for the manufacture of autologous cell therapies, driven by the need to scale-out and the potential need to manufacture therapies at the point-of-care. Comparability as an example becomes a critical translational bottleneck that is perceived by many as a ‘show stopper’ for autologous scale-out. In this specific case, the existing regulation appears to remain uncompromising with respect to the level of assurance required to demonstrate comparability after the roll-out of manufacturing processes to multiple sites post-clinical trial.
A debate is needed on the role of regulators and stakeholders in the risk/benefit decisions that surround alternative business models for MTMM autologous cell therapies, specifically concepts such as ‘GMP in a box’ (in which self-contained modules are used to prepare cell based therapeutic products close to clinic or in local production hubs similar to franchised operations) and point-of-care manufacturing. Such a debate could impact national competitive advantage (e.g. through the emergence of new regulatory paradigms such as those recently adopted in Japan, South Korea and Australia). This is not simply a matter of changing the regulations. It requires a constructive alliance between regulatory bodies, stakeholders and researchers to seek mutually acceptable and appropriate levels of confidence in production using alternative technologies and flexible controls. It does, however, reflect the need for enhanced proactivity in the stated aim of the regulator to protect, promote and improve public health.
This is an instance of a requirement for an effective pre-competitive community of practice in regulatory and manufacturing science of which comparability studies are a part and where the outcome of the scientific activity informs both the regulatory body and the manufacturer. Such work will assist the balancing of innovation for patient benefit with the management of risks to patients – the voice of industry is increasingly seeking assurance that there is balance in the application of the regulation. To this extent, the House of Lords call for a coordinated overhaul of all the processes involved with the clinical translation and commercialization of cell therapies in the UK is welcomed (HoL Report, 2013). Regulators are beginning to understand these wider roles and the responsibilities that go along with this, particularly when it comes to novel areas of innovation like Regenerative Medicine. The challenge for regulators has now become one of maintaining the safety, quality and efficacy of the products they license while also enabling innovative products and processes to be delivered to healthcare markets in the public or commercial sectors on a shorter timescale and with less cost. Regulatory science is part of this agenda.
Acknowledgements
This review was based on work conducted as part of the ‘Exploring the Feasibility of a New Regulatory Paradigm for the Manufacture of Autologous Cell Therapies’ project funded by EPSRC Centre for Innovative Manufacturing in Regenerative Medicine. The authors declare no conflicts of interest
References
Last revised March 31, 2014. Published June 30, 2014. This chapter should be cited as: Hourd, P., Chandra, A., Medcalf, N. and Williams, D. J., Regulatory challenges for the manufacture and scale-out of autologous cell therapies (June 30, 2014), StemBook, ed. The Stem Cell Research Community, StemBook, doi/10.3824/stembook.1.96.1.