
The cell therapy industry is rapidly growing with several new products approved for clinical use over the past few years and many more currently in clinical trials. Nonetheless, a number of challenges remain in getting cell therapies to the market and into routine use. For instance, cell therapy bioprocesses still need to be optimised, but if these therapies are to be widely adopted there must also be clear routes for their delivery from the site of manufacture to the clinic and the patients. This review explores the currently available routes, the factors that need to be taken into consideration when choosing a route and the challenges that remain with respect to cell therapy logistics.
1. Introduction
The term “cell therapy” encompasses a range of activities including; therapies that look to either permanently or transiently replace cells, are immunomodulatory or use the cells as delivery vehicles for other technologies such as gene therapy (Mason et al., 2011). At its heart is the use of either the patient's own cells (autologous therapies) or donor cells (allogeneic therapies). Said to be the fourth therapeutic pillar of healthcare (Mason et al., 2011) and with induced pluripotent stem cells touted to be “a game changer” (Inoue et al., 2014), the cell therapy industry continues to grow and is estimated to reach an annual turnover of $20bn worldwide by 2025 (Cell Therapy Catapult website). Recent years have seen clinical trials show signs of success (Ratcliffe et al., 2013a) and products such as Prochymal (Osiris Therapeutics Inc) and ChondroCelect (TiGenix) gain regulatory approval. However, many new cell therapies are treating relatively small numbers of patients so a universal system for delivery of these products from the site of manufacture to the clinic has not yet been established. This review explores whether a universal system is realistic and explores the options for delivery of a cell therapy product to the clinic and identifies factors that need to be taken into consideration when deciding on a route. Importantly, it also highlights some of the challenges that still face the industry in this regard which need to be tackled if these therapies are to be adopted more widely.
2. Possible delivery routes
Often identified as a peripheral, rather than core, element of cell therapy bioprocessing (Ratcliffe et al., 2011), cell preservation nonetheless plays a key role in the manufacture of these therapies, enabling the generation of cell banks and transport of either cells and/or the final product. Indeed, one of the oldest examples of a cell therapy, the transfusion of red blood cells in cases of severe anemia, relies on refrigerated storage. Red blood cell units are typically donated, transported to one of several national blood banking facilities, processed and then preserved within an additive solution to give a shelf-life of up to 42 days (Hess, 2012). They are subsequently transported to hospitals where their storage and use is closely monitored to manage waste and comply with regulations.
However, alternative routes into the clinic, depending on site of manufacture and method of preservation are possible as shown in Figure 1. For example, cells could be expanded, purified and cryopreserved into vials or bags at a manufacturing site before being shipped to the hospital, stored and processed and, ultimately, delivered to the patient. The extent of that final processing will depend on the product itself and is discussed further in the ‘Can the cell therapy be processed at the clinical site?’ section. Indeed, many existing cell therapy products and those in clinical trials are cryopreserved, including Prochymal and other mesenchymal stem cell (MSC)-based products (Ikebe & Suzuki, 2014; Kebriaei et al., 2009; Mendicino et al., 2014). Alternatively, some therapies, such as haematopoietic stem cell (HSC) transplantation or ReCell (Avita Medical) can avoid prolonged cell preservation and transport altogether, administering freshly isolated and processed cells to the patients within a few hours (Gilleard et al., 2013; Giralt and Bishop, 2009).
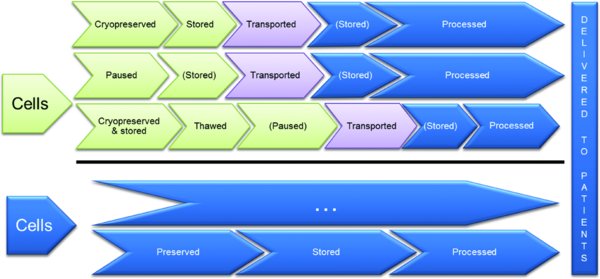
Cells, autologous or allogeneic, are first processed (this could include thawing from a cell bank or isolation from a fresh biopsy, activation and/or expansion) before being subsequently preserved (cryopreservation or under hypothermic conditions, i.e. ‘pausing’) or delivered directly to patients. Stages marked in blue are carried out at the clinic, those in green at a separate manufacturing site and purple indicates transportation is required. Stages in brackets are optional. Critical elements such as product release testing have not been included here for simplicity.
3. Deciding which route is suitable for a product
The choice between the routes outlined in Figure 1 will depend on a number of linked factors such as: whether the product is autologous or allogeneic, how it is manufactured and if/how it is preserved for storage and transportation. Below are some of the key questions that need to be considered for any given product. Importantly, these must be considered early on in its development to ensure that processes do not need to be changed and revalidated to account for scale if the therapy is successful in clinical trials.
3.1. Can the cell therapy be manufactured at the clinical site?
Autologous therapies, such as HSC or MSC transplantation, can lend themselves to manufacture at the clinic where all processing from initial cell harvesting to final delivery to the patient is done on-site. Therefore, suitable facilities and specially trained staff are required that could, depending on the therapy, isolate the cells of interest from the starting material, expand, characterise and formulate them into the final product. Devices such as Cytori's ‘Celution System’ (Lin et al., 2008), which has recently been approved for use in Australia to process and deliver adipose-derived regenerative cells (Cytori website), can overcome some of this burden and enable point-of-care manufacture. However, depending on how a product is classified from a regulatory perspective, there may be an associated need to meet GMP and quality assurance requirements (Table 1). As many clinical sites are not working to GMP this is a major hurdle and thus some autologous therapies are being manufactured at central facilities. For example, ChondroCelect, an autologous treatment of cartilage cells for knee cartilage defects is manufactured at TiGenix in the Netherlands, a process which takes at least 4 weeks (ChondroCelect: EPAR-product information). Similarly, Provenge (Dendreon), an autologous treatment for certain forms of prostate cancer is manufactured by PharmaCell in the Netherlands but over no more than 3 days (Provenge: EPAR-product information).
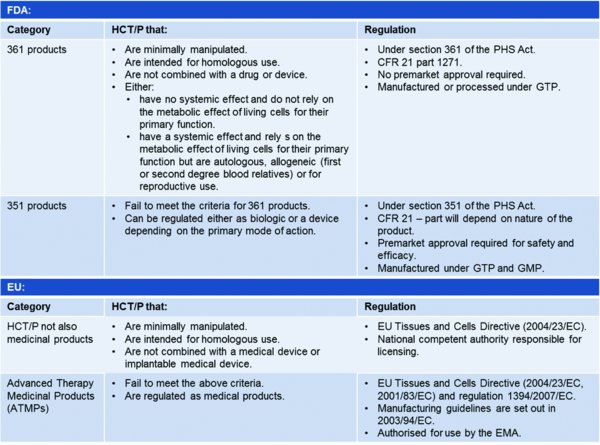
(PAS83:2012; Thirumala et al., 2013). Notably, a limited number of cell therapies, including blood derived products and minimally manipulated bone marrow for homologous use are not regulated under CFR 21 in the USA. For more information on what is considered to be a minimal manipulation or how homologous use is defined, please refer to PAS83. CFR: Code of Federal Regulations, EMA: European Medicines Agency, HCT/P: human cells, tissues, and cellular and tissue-based products, PHS: Public Health Services.
Clearly, with allogeneic therapies where cells from a single donor are used to treat many patients, larger scale bioprocesses can be established that are streamlined, potentially automated and more economical (Brindley et al., 2013; Ratcliffe et al., 2011). However, initial capital expenditure to set up a single manufacturing facility will be high. For instance, a manually-operated roller bottle-based system of 1500 bottles per lot to generate a virus product is estimated to cost approximately $58M although there is a cost benefit to introducing automation (Brindley et al., 2013; Dutton & Fox, 2006).
An alternative strategy, which has the potential to benefit businesses and clinics alike, is to adopt a distributed manufacturing business model. An example of this approach can be summarised in the following way: consider a business that delivers a mid-range value allogeneic therapy using anchorage-dependent cells at high volume (i.e. not a therapy for a life-threatening condition, but a remedial therapy for a moderately disabling condition with a global market). The simple calculation of the cells required may show that the growth surface required for the cells per annum mandates a very large facility if traditional disposable technologies, such as cell factories, roller bottles or T-flasks, are to be used (Simaria et al., 2014; Want et al., 2012). The choice of such methods is attractive in the sense that the growth environment is comparable to that which was applied during early development and proof-of-concept. However, the business then faces a dilemma: should it invest in an alternative, more intensive method of production before proceeding with clinical trials (thus incurring a delay to launch and additional technical risk) or should it absorb the large capital cost of building a factory for an as yet unproven sales projection. If the latter choice is made then investment capital may be hard to find and operability problems will be incurred when manipulating such disposables at high volume. The dilemma can be avoided if the business opts to derogate manufacture of the ‘drug product’ (i.e. the cells in the final dosage form) to regional or local centres under a different business team. In this case the role of the central operation will be to make a more modest number of kits and provide equipment and training packages that will enable operators to make small-to-medium-sized lots of the final product in the ‘distributed’ microfactories. One advantage of this is also that it can overcome some of the issues surrounding long-distance travel with short shelf-life products and delays at customs. However, at present this approach is very challenging due to the requirement to manage the issue of batch-to-batch comparability between sites. There remain unanswered questions about the definition of the batch identity, process capability and the ownership of ultimate responsibility for placing the goods on the market. Automated microfactories with minimal human intervention may hold the key that will unlock this option. These questions will, if addressed well and responsibly, open up a new paradigm of operation for otherwise difficult to manufacture products.
3.2. Can the cell therapy be processed at the clinical site?
Regardless of whether a cell therapy is manufactured at a large single facility or microfactories, the final product may still require some processing before administration to the patient. The nature of this processing depends on the product and how it has been preserved but could include thawing, washing to remove the protective agents used in cryopreservation (Calmels et al., 2003), dilution to target cell number (Kebriaei et al., 2009) or simply resuspension. Shelf-life assays need to take into account the time required for these and any temperature or other environmental shifts encountered to ascertain their impact on the final product. Furthermore, suitable facilities at the clinical site and/or bedside will be needed and staff must be trained in how to carry out these procedures. The choice of preservation method, and any subsequent post-preservation processing it necessitates, may therefore play a role in how readily or widely adopted a therapy can be. For example, ChondroCelect is stored under ambient conditions and can be implanted into a cartilage defect after simply resuspending the cells by tapping the vial (ChondroCelect: EPAR-product information), negating the need for any specialised equipment. In contrast, Provenge only has an 18 hour shelf-life once in its insulated container and when removed, must be fully infused into patients within 3 hours (Provenge: EPAR-product information) making careful coordination of patient, clinician and shipping necessary.
The responsibility for product release to the patient is especially important and must be managed with care. For centralised manufacture the principles of GMP require that batch testing to specification is conducted and the results, combined with a review of the production records and the results of inspections of the facility and the operation, be reviewed by the Quality Unit before authorization is given to release the batch (21 CFR 211.65(a)). However, under certain circumstances where the therapeutic value of the product will be lost if there is significant delay in release pending the execution of longer tests, such as sterility testing, the batch may be released ‘at risk’ for a well-controlled process, provided always that any outstanding tests are completed as soon as possible.
In the case of many new autologous therapy processes the products are made on a ‘Hospital Exemption’ basis and the principles of GMP are side stepped. However, for a regulated autologous product there is a challenge for control of quality in the form of timeliness of product tests and the quantity of the (usually small) batch required for testing. The potential for monitoring and control data, preferably from a non-destructive at- or in-line measurement system, to provide in real time the majority of the information needed for product release is expressed in the FDA's guidance on Process Analytical Technologies (PAT; James, 2011). If the product were to be supplied from a central factory then the responsibility for release would rest clearly with the central manufacturer. For goods made and applied on an exemption or named-patient basis the quality unit at the hospital or clinic would make the decision under written protocols. However, for a distributed manufacturing model of supply the situation is less clear. It is possible to envisage mechanisms for local automated manufacture coupled with relay of PAT data back to the central, supporting company permitting the central business to make the release decision. Such a situation has yet to be established in practice. At present, hospital roles with respect to release are restricted at one extreme to dispensary staff inspection of purchased goods for correct labelling, package integrity and conformity to the expiry date criterion and at the other extreme to viability testing and confirmation of asepsis for treatments such as pancreatic islet cell transplants supplied from another centre.
3.3. Can the cell therapy be transported?
Transport and temperature controlled container systems for a variety of therapies from traditional pharmaceuticals through to organ transplants and blood products already exist, however, cost and transport delays (including through customs) can be an issue. Notably, good distribution practice guidelines highlight that “regardless of the mode transport, it should be possible to demonstrate that medicines have not been exposed to conditions that may compromise their quality and integrity” (Good distribution guidelines). This includes monitoring elements such as the temperature fluctuations that a vial or bag of product experiences during transit to check, before the product is administered, that this stayed within defined limits. The setting of those limits presents a problem, as they must be broad enough to allow the desired transfer to occur, but not so much that the quality of the product becomes compromised. Unfortunately, the issue is further compounded by our current lack of understanding of how cell therapy ‘quality’ should be defined. For example, the mode of action of some cell therapies is unclear, therefore measurement of cell quality or product potency is often ill-defined and reliant on expression of non-specific protein markers. It is important that these issues are resolved, because without a potency assay cell therapy shelf-life cannot be determined. Thus, the International Society for Cellular Therapy recently released a comprehensive review of some of the challenges the industry faces with respect to these issues and provides some case studies where they have been overcome (Bravery et al., 2013). Notably, although red blood cell transfusion has been used for decades, it is known that a number of molecular changes, so called ‘storage lesions’ can occur within the cells. Although some, such as haemolysis, have an obvious effect on clinical function, for others, the link to clinical outcome is less conclusive, calling into question what the minimum release criteria for units of red blood cells or regulatory testing for new additive solutions should be (Hess, 2014). This indicates that the debate over how to measure product quality in manufactured cell therapies is also unlikely to end soon.
It may also be incorrect to assume that temperature alone has an impact on the cells. Mechanical forces have been shown to affect MSC differentiation potential (Wang and Chen, 2013) and Nikolaev et al., (2012) showed that under hypothermic conditions, human MSCs were sensitive to vibrations that mimicked road transportation of cells suspended in vials. They reported that cells that had already been stored at 4ºC for up to 144 hours experienced a greater reduction in cell viability when exposed to vibrations than those exposed immediately after being suspended. Whilst this study was rather limited because the MSCs were stored in simple culture medium rather than tailored hypothermic storage medium, it highlights that the effect of transportation as a whole on cell therapy products should be further investigated.
3.4. Can the cell therapy be preserved and if so, how?
The ability to preserve cell therapies is desirable from a number of perspectives. It gives these products, whether they are autologous or allogeneic, a shelf-life, thereby enabling them to be manufactured off-site. For example, Prochymal, an allogeneic, bone-marrow derived MSC product which has been approved for use in the management of acute GvHD in paediatric patients in Canada, has a shelf life of 2 years at ≤–135ºC (SBD for Prochymal). Preservation allows for patients to be treated multiple times with the same batch of cells if, for example, a graft fails. It also enables cells to be harvested for later use, a fact exploited in the cord blood banking industry. Importantly, it also enables greater flexibility in therapy timing as treatments consisting of freshly isolated and processed cells put pressure on patients, donors and staff as timing of cell, patient, theatre and clinician availability is critical. Furthermore, preservation enables autologous cells that will not be expanded, to be harvested over a number of sessions, or days, if insufficient cells are harvested in a single procedure. For example, in trials with Provenge, it was found that in approximately 25% of cases, more than 3 leukapheresis procedures were required in order to generate the cell doses required for treatment (Provenge: EPAR-product information).
There is a wealth of information and large range of protocols available regarding the preservation of cells, including stem cells (the reader is directed to a number of recent reviews: Coopman, 2011; Golab et al., 2013; Hunt, 2011; Robinson et al., 2014; Thirumala et al., 2013). A recent overview of proposals submitted to the FDA for MSC-based products indicated that >80% of 66 submissions considered used cryopreservation to store and transport their product (Mendicino et al., 2014). It has even been suggested that cryopreservation may improve the clinical efficacy of Dermagraft (Naughton and Applegate, 2002). Yet cell preservation has typically been linked to significant cell losses and in some instances, changes in cell phenotype and function and/or clinical outcome. For example, protocols have been developed that enable the survival of human hepatocytes after cryopreservation but a large proportion of cells lose their ability to attach to surfaces, which is key for engraft, and their metabolic function is altered (Terry et al., 2006). Similarly, freshly thawed MSCs showed significantly reduced viability (as low as 44%) as well as a decreased ability to suppress T-cell proliferation in vitro and respond to IFNγ compared to actively growing MSCs (François et al., 2012). Furthermore, the debate on the impact of cryopreservation on allogeneic HSC transplant engraftment and risk of GvHD continues (Frey et al., 2006; Parody et al., 2013). Unsurprisingly, thawed MSCs that were cultured for 24 hours regained these immunomodulatory–linked functions (François et al., 2012) and thawed T regulatory cells that are cultured post thaw also regained their suppressive activity in vitro compared with freshly thawed cells (Peters et al., 2008). This indicates that a period of cell ‘recovery’ post-thaw should be factored into cell therapy administration to optimize cell function. However, the benefit of this recovery to clinical outcome compared with the cost of the required processing facilities would need to be considered and further studies done to ensure that any recovery period is not selecting for, or enabling expansion of, culture adapted undesirable cells.
Cells are typically cryopreserved in a medium containing DMSO or other compounds, so-called cryoprotective agents (CPAs), which protect cells from some of the damage caused during freezing. However, these CPAs are typically toxic to cells at temperatures above 0ºC in a time and concentration dependent manner. Furthermore, some adverse reactions in patients have been recorded when a thawed cell suspension including DMSO is infused (Cox et al., 2012; Shu et al., 2014). Although not all adverse reactions have been directly attributed to DMSO (Shu et al., 2014) and the impact on cell quality of post-thaw exposure to DMSO has not been studied, washing protocols to remove CPAs and cell debris from thawed cell therapies prior to administration have been developed. The main downside to these is that varied cell losses have been reported (Calmels et al., 2003; Fleming and Hubel, 2006; Rodriguez et al., 2005; Sánchez-Salinas et al., 2012; Shu et al., 2014). For example, Calmels et al. (2003) reported that when the CytoMate (Baxter) was set up to remove 96% of DMSO up to 40% of cells were lost. One study also indicated that the inclusion of washing step delayed platelet recovery in patients receiving a peripheral blood progenitor cell transplant (Akkök et al., 2009) highlighting the impact of post-thaw processing, and not just cryopreservation, on cell survival and/or clinical outcome.
In a recent review into the failure of the Phase III trial of Prochymal for steroid—resistant GvHD to meet its primary clinical endpoint, the author proposed that its formulation as a cryopreserved product may have contributed (Galipeau, 2013). In particular the author highlighted the discrepancy between pre-clinical studies of MSCs in animal models of human disease where live, actively growing cells are administered, compared with clinical trials using freshly thawed cells where the only release criterion is often that cell viability by trypan blue exclusion is >70%. This calls into question not only how pre-clinical studies are performed but also whether viability by trypan blue exclusion is sufficient as a release criterion. The latter is particularly questionable given that trypan blue can overestimate the number of viable cells compared to other methods (François et al., 2012; Altman et al., 1993, Mascotti et al., 2000) and cryopreservation may cause delayed onset apoptosis (Van Buskirk et al., 2005) which will not be detected by trypan blue.
Hypothermic preservation, where cells are maintained above 0ºC, is an alternative to cryopreservation for the short-term preservation of cells because cell metabolism is significantly slowed so that they are effectively ‘paused’. Within the pharmaceutical industry this can be used to uncouple cell culture from cell-based assays (Wise et al., 2009) and in the manufacture of recombinant proteins, mild hypothermia (25--33ºC) can be used to improve product yield (Roobol et al., 2009). This concept is also not new to the cell therapy industry, because products such as red blood cells have long been transported and shipped under refrigerated conditions and freshly isolated allogeneic bone marrow or peripheral blood HSC products can be stored at 4ºC for 24-48 hours prior to infusion (Andritsos et al., 2007). More recently, both ChondroCelect and Carticel (Genzyme) are formulated in DMEM and have a shelf-life of just 48 and 72 hours, respectively when kept under the appropriate storage conditions (Carticel packing insert; ChondroCelect: EPAR-product information). Similarly, Provenge is formulated in simple Lactated Ringer's Injection solution but only has a shelf-life of 18 hours in its shipping container and must be used within 3 hours once removed from that container (Provenge: EPAR-product information). In 2012, TiGenix also completed the first phase I clinical trial with cells stored and administered in HypoThermosol (BioLife Solutions; NCT01743222), one of several specialised hypothermic storage media available on the market. However, the data publically available on hypothermic storage of cells, whether at 2--8ºC or ambient temperatures, remains limited (Robinson et al., 2014). In particular, despite the fact that cells for cell therapy will more than likely be stored in suspension, most research is focused on how cells adhered to a surface respond to hypothermic conditions, limiting the relevance of these studies. Nonetheless, they do highlight that, as with cryopreservation, it may not be possible to establish a platform process for hypothermic storage as studies have shown adaptation of basic hypothermic medium may be needed to ensure cell survival. For example, Matthews and colleagues found that HypoThermosol supplemented with Trolox and RGD peptide was the best media for storage of skeletal muscle cells at 4ºC, whereas hepatocytes required the inclusion of a caspase inhibitor, FK041 (2004).
As with cryopreserved products it will also be important to understand what impact, if any, hypothermic storage has on cell quality. For example, Ostrowska and colleagues reported that although the viability of human hepatocytes stored in HypoThermosol-FRS at 4ºC for up to 72 hours was high (∼70%), the metabolic function of the cells was approximately half that of freshly isolated cells (Ostrowska et al., 2009). It may be that ‘paused’ cells would also benefit from a period of recovery after rewarming.
Notably, cryopreservation and cell pausing can also be used in conjunction with one another, and not just in the generation of allogeneic treatments where master cell banks are generated. For example, in the manufacture of Carticel, autologous cultured chondrocytes for the repair of cartilage defects at the femoral condyle, cells are initially isolated from a biopsy, cultured and frozen. They are then thawed at the manufacturing facility, further expanded and finally shipped to the clinic once the implantation has been scheduled (Carticel website). This gives the patients and clinicians greater flexibility compared to, for example, Provenge, where a strict schedule of apheresis and transfusion procedures must be adhered to.
4. Future perspectives
The cell therapy industry is rapidly growing with several new products having been approved for clinical use over the past few years and many more in clinical trials at the time of writing. There are many challenges that need to be tackled before cell therapies are widely adopted, including the optimization of their manufacture. With much of the research focus on how cells can be effectively isolated and expanded, relatively little attention has been given to how these cell therapies will be preserved and physically transported to the clinics and patients. Whilst a single platform process or system is unlikely to suit the industry given the disparate nature of cell therapy products, several universal challenges and questions remain which are summarized in Figure 2.
Improved preservation processes that minimize cell loss post-thaw or –rewarming and are compatible with mode of administration to the patient (e.g. implantation or infusion), are still needed. Typically, the impact of changing a single process parameter such as CPA choice or concentration on survival of cells in cryovials is investigated. Hence, the relationship between parameters such as cryoprotective agent choice, cell density, equilibration times, freezing rate, vial/bag dimensions and so forth are poorly understood if at all. Use of multifactorial design of experiments or quality by design approaches, such as that employed by Ratcliffe and colleagues in the optimization and validation of a single subprocess in the manufacture of human embryonic stem cells (Ratcliffe et al., 2013b), will help optimize preservation processes.
Consideration must also be given to the shelf-life and stability of cell therapy products and how they should be stored and/or processed at the clinical site. Despite the efforts that have gone into defining freezing rates and profiles for cells and developing controlled rate freezers that can achieve these reliably, vials of cells are typically thawed without any control and the impact of this on cell quality or clinical outcome has not been systematically explored. The need for low temperature storage and short shelf-lives currently also limits the applicability of cell therapies in, for instance, war zones. Research into the potential to lyophilise red blood cells so they could be stored and shipped under ambient conditions is ongoing but efforts to desiccate nucleated cells have been limited (Coopman, 2011). Nonetheless, this is an exciting area of research that would represent a real step-change in how these therapies are delivered.

In addition, more work is needed to continue developing cost effective low temperature storage solutions that will help maintain conditions if cell therapy products are detained at customs or need to travel further if distributed manufacturing is not feasible. Importantly, as this review has highlighted, how and where a product is manufactured, how it is preserved and how it is transported are all inextricably linked and can impact product quality. Thus, how a product is to be delivered to the clinic must be taken into consideration during its development to ensure that, if successful through clinical trials it can be adopted more widely.
Acknowledgements
The authors would like to acknowledge the Biotechnology and Biological Sciences Research Council (BBSRC; UK), Bioprocessing Research Industries Club (BRIC) and the Engineering and Physical Sciences Research Council (EPSRC; UK) for their financial support.
References
Last revised March 31, 2014. Published June 30, 2014. This chapter should be cited as: Coopman, K. and Medcalf, N., From production to patient: challenges and approaches for delivering cell therapies (June 30, 2014), StemBook, ed. The Stem Cell Research Community, StemBook, doi/10.3824/stembook.1.97.1.