
The prospect of personalized regenerative medicine promises to provide treatments for a wide range of degenerative diseases and medical conditions. An important first step in attaining this goal is the production of pluripotent stem cells directly from individual patients, thereby providing autologous material which, after correcting intrinsic genetic defects and differentiation into required cell types or tissues, could be transplanted into the patient. This chapter reviews the current progress towards this first step, focusing on the techniques used to generate pluripotent cells, the advantages that each offers and the challenges that must be overcome.
1. Introduction
Much of the hope invested in embryonic stem (ES) cell research surrounds its promise to provide a broad spectrum of medical applications. The development of such treatments relies on the production of pluripotent stem cells genetically identical to patients. Such stem cells, following differentiation into the disease-relevant cell types, would serve as the key substrate for disease models to study the patient's condition, drug discovery to slow or stop cellular degeneration, and cell replacement therapies after any intrinsic disease-causing genetic defects were repaired (see Figure 1). Beyond providing a renewable source of material that might be transplanted into a patient without suppressing their immune system, the generation of autologous pluripotent stem cells provides two key advantages over the large number of existing stem cell lines. First, evidence exists that off-target effects of immunosuppressant drugs commonly used in tissue and organ transplants may directly interfere with the function of transplanted tissues, such as the inhibition of β-cell replication that has been observed with drugs used in the treatment of Type I Diabetes (Nir et al., 2007).

The production of cellular therapies requires the optimization of four steps: first, isolating and culturing cells that can be readily obtained from a patient in a non-invasive fashion. Second, the reprogramming of these cells into a pluripotent state. Third, the directed differentiation of those patient-specific pluripotent cells into the cell type relevant to their disease. And, fourth, techniques for repairing any intrinsic disease-causing genetic defects and transplantation of the repaired, differentiated cells into the patient. Notably, these disease-relevant patient cells can also be used for in vitro disease modeling which may yield new insights into disease mechanisms and drug discovery.
The second advantage of autologous ES cells over existing lines concerns the ability to create effective disease models. While some success has been reported recently in the in vitro modeling of degenerative diseases by introducing known disease-causing alleles into ES cells (Di Giorgio et al., 2007; Nagai et al., 2007; Yamashita et al., 2006), or by using ES cell lines established from embryos following preimplantation genetic diagnosis (Eiges et al., 2007), such models are severely limited in their scope as they can only investigate the mechanisms of diseases for which causal genetic liaisons have been identified. Unfortunately, however, this is not the case for many conditions. For instance, fewer than 10% of patients with Amyotrophic Lateral Sclerosis (ALS, also know as Lou Gehrig's Disease) suffer from a familial form of the disease with a known genetic correlate (Dunckley et al., 2007). It therefore remains unclear whether recent progress with models studying a handful of known disease-causing alleles is applicable to the vast majority of patients with sporadic forms of this degenerative condition. Disease models using pluripotent cells generated directly from patients with these sporadic conditions could directly address such questions, as well as provide new insights into the mechanisms and progression of such syndromes.
This chapter focuses on the methods and techniques available for the production of patient-specific pluripotent cells, the promises and limitations of each technology, and a discussion of the current progress towards this goal. Several techniques have been described for restoring developmental potential to a terminally differentiated nucleus (see Figure 2). These include: nuclear transfer, wherein the chromatin of an oocyte or zygote is replaced with that of a somatic donor cell; fusion of somatic cells to ES cells; cell-culture-induced reprogramming after explantation of tissue from neonatal or adult testes; and the retroviral-mediated introduction of a small number of genes known to play a role in pluripotency. The advances, breakthroughs, and challenges surrounding each of these methods are discussed in detail below.

Four techniques for restoring developmental potential to a somatic nucleus have been described in the literature. In nuclear transfer, the genetic material of an oocyte or zygote is replaced with that of a differentiated cell such as a fibroblast. Following development to the blastocyst stage, pluripotent ntES cells can be derived as from fertilized embryos. In cellular fusion, hybridization between ES cells and somatic cells yields tetraploid ES cell lines. In direct reprogramming, the retroviral-mediated introduction of a small number of transcription factors is sufficient to confer a pluripotent phenotype. Finally, explantation of testes tissue from neonatal and adult mice into appropriate culture conditions has been shown to result in the production of multipotent adult spermatagonial (MAS) cells.
2. Nuclear transfer
Building on the early work of developmental biology pioneers such as Spemann (Gurdon and Byrne, 2003; Spemann, 1938), nuclear transfer (NT; also commonly called somatic cell nuclear transfer, SCNT) experiments were first devised in the 1950s as a means to investigate the constancy of the genome: that is, whether cells maintained the full complement of genomic information as they became more differentiated. At the time, many believed that each cell fate decision during development involved the progressive loss of genes that would not be used by the more differentiated progeny. For instance, ectoderm precursors eliminating all endoderm- and mesoderm-specific genes, then eliminating skin-specific genes as the decision to become a neural precursor is made, eventually yielding a specific type of neuron with a minimal genome containing only the genes which would actually be transcribed. NT experiments in the frogs Rana pipiens by Briggs and King (Briggs and King, 1952) and Xenopus laevis by Gurdon (Gurdon et al., 1958; Gurdon et al., 1975) indicated that, although the generation of clones became less and less efficient as the developmental age of the donor nucleus increased, it was possible to obtain heartbeat-stage tadpoles from terminally differentiated adult cells. It was not until the more recent cloning of Dolly the sheep (Wilmut et al., 1997), however, that researchers succeeded in using a cell from an adult animal to generate another healthy, fertile adult, thereby demonstrating that the nuclei of at least some cells in the adult maintained a full developmental capacity. While an important finding, many speculated that the rare cloned adult animals could have arisen from the nucleus of an equally rare somatic stem cell. Definitive demonstration that terminally differentiated adult nuclei maintain full developmental capacity was later achieved in mouse studies using mature lymphocytes (Hochedlinger and Jaenisch, 2002; Inoue et al., 2005) and olfactory neurons (Eggan et al., 2004) as NT donors.
Dolly's birth and an initial report of the derivation of human ES cells from discarded in vitro fertilization (IVF) embryos shortly thereafter (Thomson et al., 1998) led to wide speculation in both the media and scientific community about the possibility of therapeutic cloning. That is, performing nuclear transfer with a patient's somatic cells to generate a preimplantation embryo from which patient-specific ES cells might be derived for use in personalized regenerative medicine. With this motivation, several studies in mouse have sought to compare the properties of SCNT-derived ES (ntES) cells with those derived from naturally fertilized embryos (Brambrink et al., 2006; Wakayama et al., 2006), as well as show proof-of-principle demonstrations of these cells’ utility in regenerative medicine (Barberi et al., 2003; Rideout et al., 2002; Tabar et al., 2008). The equivalence of ntES cells to ES cells from fertilized embryos was of particular concern because of the low efficiencies and common health defects associated with animals brought to term after reproductive cloning (Eggan et al., 2001; Gurdon and Byrne, 2003; Humpherys et al., 2001). Despite these defects, however, two studies evaluating the transcriptional profiles, DNA methylation patterns, and in vitro differentiation capacity found that mouse ntES cell lines were identical in all regards to genetically-matched control ES cell lines derived after fertilization (Brambrink et al., 2006; Wakayama et al., 2006). These reassuring results, paired with reports using ntES cells to treat mouse models of both severe compromised immunodeficiency (Rideout et al., 2002) and Parkinson's diseases (Barberi et al., 2003; Tabar et al., 2008) after in vitro genetic manipulations and differentiation into the required cell types, generated immense hope that applications to human diseases were immediately on the horizon.
While ntES cells hold great promise for the field of regenerative medicine, the technique has several significant drawbacks that hinder its potential for widespread application to medicine or even to the study of the process of nuclear reprogramming. Obstacles associated with the requirement for a scarce and politically charged cell type, human oocytes, as a recipient cytoplasm are only amplified by the extreme technical challenge of a method plagued with inherent inefficiencies. Moreover, the experimental requirements for successful nuclear transfer in primates appear to be considerably different than other species, with attempts to apply the methods and techniques from mouse directly to human thus far yielding nothing more than false starts (Kennedy, 2006) and the rare report of NT-derived blastocysts but with no ES cell lines derived from them (French et al., 2008; Stojkovic et al., 2005). Likewise, only very recently has successful nuclear transfer in monkeys been reported (Byrne et al., 2007), but the technical refinements developed here have failed to immediately translate into advances with human cells.
A particularly severe limitation of human therapeutic cloning which has significantly hindered researchers’ capacity to develop the technique is the difficulty in obtaining donated oocytes. A 2007 study in mouse suggests that there may be alternative sources for the recipient cytoplast (Egli et al., 2007). While early mouse NT studies in which the enucleated interphase zygote was used as a recipient lead to the conclusion that reprogramming capacity was lost following fertilization (McGrath and Solter, 1984; Wakayama et al., 2000), Egli and coworkers demonstrated that by removing the chromatin from a zygote arrested in metaphase just prior to the first cell division and introducing the chromatin from a metaphase-arrested somatic cell into this cytoplast, reprogramming could occur with success rates comparable to NT into the mature oocyte (which is naturally arrested in metaphase; see Figure 3). The authors reasoned, therefore, that nuclear factors, trapped in the interphase nucleus but present in the cytoplasm during metaphase due to nuclear envelope breakdown, were necessary for reprogramming to occur. Not only might the metaphase zygote therefore serve as an appropriate recipient for NT, but this result suggests that other cleavage-stage blastomeres, arrested in mitosis, may be as well. These findings both shed some mechanistic light on the process of reprogramming and open the door to using a wider range of materials for human NT experiments. Although on the surface this study holds great promise for human NT experiments, it is unfortunately not common IVF practice to freeze or discard zygotes or early cleavage-stage embryos, as embryo quality can be difficult to assess so soon following fertilization (Salumets et al., 2001). Of particular interest, however, Egli et al. further demonstrated that polyspermic zygotes (that is, embryos fertilized with multiple sperm) could be used for successful nuclear transfer. Multiple fertilization is relatively common in IVF (roughly 3–5% of zygotes) and these embryos, which have no clinical use, are routinely discarded (Anon, 2004; van der Ven et al., 1985). As such, discarded polyspermic IVF embryos may present a valuable new avenue towards success in human nuclear transfer.
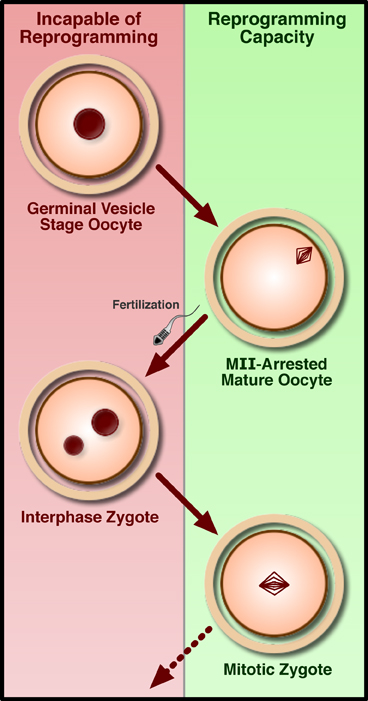
Development fails after replacing the interphase nucleus of either a germinal-vesicle stage oocyte or pronuclear zygote with somatic chromatin. However, transfer of somatic chromatin into either the MII-arrested oocyte or a zygote arrested with a drug in the first mitosis allows for the generation of cloned mice and ntES cells.
Despite the challenges and limited achievements in human, NT remains the “gold standard” in nuclear reprogramming with clear demonstrations of the production of both healthy clones and pluripotent stem cells identical to those derived from fertilized embryos. Nevertheless, the development of more robust and technically simple reprogramming methods (discussed below) appears to be at hand, leading some authors to write obituaries for SCNT (Cibelli, 2007; Highfield, 2007). Whether these eulogies are premature or whether the quality of pluripotent cells generated by newer techniques will prove to be as high as ntES is presently an area of intense investigation.
3. Cellular fusion
Fusion of various somatic cells and cell lines, generally using interspecies hybrids to distinguish genes expressed from each nucleus, has long been used to investigate phenotypic dominance at the cellular level. For instance, when fibroblasts are fused to myoblasts, are the resulting hybrids more like fibroblasts, myoblasts, or something in between? (Harris, 1965; Mevel-Ninio and Weiss, 1981; Wright, 1984). Initial results indicating that some cellular identities could dominate over others in hybrids generated hope that this system could be used to investigate the mechanisms of this fate respecification as a proxy for understanding the effectors of cell fate decisions normally made in the process of development (Blau et al., 1985; Boshart et al., 1993). However, between technical problems with interspecies fusion and the derivation of embryonal carcinoma (EC; Martin and Evans, 1975), and subsequently ES cells (Evans and Kaufman, 1981; Martin, 1981), as more accurate in vitro models for cell fate determination, work with fusion waned considerably for several decades. Interest in this line of investigation was reinvigorated following the advent of mammalian NT and the subsequent speculation about the prospects of nuclear reprogramming for regenerative medicine. The observation that a pluripotent phenotype appeared to dominate following the fusion of murine somatic cells to EC (Miller and Ruddle, 1976), embryonic germ (EG; Tada et al., 1997), and ES (Tada et al., 2003; Tada et al., 2001) cells seemed to promise that somatic-stem cell fusion might be an appealing alternative to inefficient and challenging NT. It was hoped that this system could be used for either the study of the mechanisms of nuclear reprogramming or perhaps eventually the direct production of patient-specific pluripotent stem cells. A report demonstrating that this capacity to reprogram somatic cells was conserved in human, as well as mouse, ES cells was further encouragement and represented the first demonstration of successful nuclear reprogramming of human somatic cells (Cowan et al., 2005).
Despite this excitement, the fusion of ES and somatic cells, as well as the subsequent reprogramming, has proven to be quite inefficient (Cowan et al., 2005; Hochedlinger and Jaenisch, 2006; Tada et al., 2001), limiting its usefulness in the study of the genetics and epigenetics of reprogramming. To date, therefore, investigators have focused on increasing these efficiencies by overexpressing genes already known to be important for pluripotency (Silva et al., 2006), rather than providing new insights about pluripotency and reprogramming through fusion. Moreover, the problems associated with inefficiency are only compounded by the tetraploid nature of the hybrids generated by the fusion itself. The presence of two complete genomes has severely limited the utility of this methodology for the study of reprogramming as well as presenting an enormous technical barrier to the production of autologous stem cells.
While the use of mature lymphocytes (which undergo a genetic rearrangement late in development) as the somatic fusion partner in the initial reports with mouse ES cells left no question that a terminally differentiated cell was being subjected to an ES cell environment (Tada et al., 2003; Tada et al., 2001), determination of the extent of reprogramming has been less straightforward. Notably, the inability of tetraploid cells to contribute significantly to a chimeric embryo has limited the characterization of the pluripotency of the hybrid cells to less-stringent in vitro assays (Jaenisch and Young, 2008; Nagy et al., 1990; Tada et al., 2001). Even more pressing, however, are questions regarding the state of the somatic chromatin in the hybrid. Has the somatic nucleus truly been restored to a pluripotent state, or has it simply silenced the transcription of genes specific to the differentiated state, allowing the pluripotent ES nucleus control the hybrid cell's identity? Studies have sought to address this concern by demonstrating the reactivation of the silent X-chromosome in hybrids formed with female somatic cells (Tada et al., 2001), activation of reporter genes (Cowan et al., 2005; Tada et al., 2001), the absence of appreciable DNA methylation at pluripotency-associated loci (indicating demethylation occurred in the somatic chromatin; Cowan et al., 2005), and the expression of some somatic-nucleus derived ES cell-associated genes by looking for specific single nucleotide polymorphisms (SNPs) in hybrid cell transcripts (Cowan et al., 2005; Tada et al., 2003). Although some studies have made use of relatively divergent strains of mice to facilitate these analyses (Tada et al., 2003), genome-wide allele-specific expression analysis would help to elucidate the state of the somatic nucleus, but has yet to be performed with either mouse or human hybrid ES cells.
Tetraploidy also presents the most significant hindrance to the use of fusion in generating patient-specific stem cells as elimination of the ES cell genome after reprogramming will be necessary to produce autologous cells. The requirement for an ES cell nucleus was demonstrated directly by one study which used ultracentrifugation of ES cells (prior to fusion) to separate nuclear and cytoplasmic material. Pluripotent cell lines could be generated following fusion of isolated nuclei (nucleoplasts) with somatic cells, but not following fusion to the enucleated cytoplasts (Do and Scholer, 2004). While these authors concluded that ES cell chromatin was required for reprogramming after hybrid formation, the successful NT results with metaphase-arrested zygotes discussed above (Egli et al., 2007) raises the possibility that mitotically arrested ES cell cytoplasts might also be capable of inducing a pluripotent state on somatic chromatin and may present a worthwhile avenue for future study. In addition to attempts to mechanically eliminate the ES cell chromatin with ultracentrifugation, a genetic system aimed at the same goal has also been developed, but to date only the elimination of a single chromosome has been demonstrated (Matsumura et al., 2007). This system, depicted in Figure 4, makes use of Cre-mediated DNA recombination between sister chromatids to generate abnormal chromosomes which are eliminated during cell division. Although promising in principle, it remains unclear whether this technique could be used to simultaneously remove the entire ES-cell genome without introducing widespread genomic rearrangements and instability. Without an effective strategy for the disposal of the ES cell's genetic material, it may never be possible to use fusion to produce therapeutically relevant stem cells, nor for that matter, even to determine unambiguously whether the somatic chromatin has been fully reprogrammed.

A “Chromosome Elimination Cassette” (CEC) consisting of two oppositely-oriented LoxP sites flanking a GFP transgene is introduced into the ES cell genome in a single copy. Following DNA replication, the introduction of Cre mediates recombination between the CEC's on the two sister chromatids, yielding abnormal chromosomes with either no centromere (nullicentric) or two centromeres (dicentric). During cell division, these abnormal chromosomes are naturally eliminated, thereby removing the ES cell-derived chromosome from the hybrid cell.
4. Cell explantation
The derivation of pluripotent ES and EG cells from mammalian embryos and EC cells from tumors is well-established, but until very recently there was little evidence for the generation of pluripotent cells directly from post-natal or adult mammals by simply explanting cells into appropriate culture conditions. Initial reports of the discovery of multipotent adult progenitor cells (MAPCs) in the bone marrow (Jiang et al., 2002) and the transdifferentiation of adult somatic stem cells (Krause et al., 2001; Lagasse et al., 2000) have to date proven refractory to independent verification, perhaps a reflection of the low stringency of analysis with which such claims of pluripotency have often been evaluated (Jaenisch and Young, 2008; Wagers et al., 2002). For instance, the mere expression of “marker” genes for different lineages does not necessarily reflect a functional demonstration of pluripotency, and is thus insufficient to justify a claim of pluripotency. A particularly noteworthy example is the recent finding that three ubiquitously-used and widely accepted markers of neuronal lineages – nestin, NeuroD1, and beta-III-tubulin – can be activated in response to cellular stress without corresponding changes in cell fate or identity (Croft and Przyborski, 2006; Neuhuber et al., 2004). Even in the very limited instances where a novel cellular function was demonstrated following a claim of transdifferentiation or culture-induced reprogramming (Lagasse et al., 2000), subsequent studies found that results were better explained by fusion of donor cells with cells in either recipient animals (Vassilopoulos et al., 2003; Wang et al., 2003) or co-culture conditions (Terada et al., 2002; Ying et al., 2002).
Although these early reports proved erroneous, a series of three recent studies indicates that pluripotent stem cells can be derived directly from neonatal (Kanatsu-Shinohara et al., 2004), and adult (Guan et al., 2006; Seandel et al., 2007), testes following cell explantation and culture in the presence of appropriate growth factors. Reasoning that EG and EC cells originate from germ cells in the embryo, and noting that some authors have suggested ES cells may have an origin in very early primordial germ cells (PGCs; Zwaka and Thomson, 2005), the germline seemed the most reasonable place to look for a conserved post-natal ability to give rise to pluripotent cells. These cells, termed multipotent adult germline stem cells (maGSCs; Guan et al., 2006) or multipotent adult spermatogonial-derived stem cells (MASCs; Seandel et al., 2007), share many of the hallmark properties of ES cells including marker expression, multilineage differentiation, and even contribution to germline chimeras in some cases (Guan et al., 2006; Kanatsu-Shinohara et al., 2004).
A key difference, however, between adult testes-derived pluripotent cells and ES cells is the status of their epigenetic imprints. Beginning with PGCs and throughout the process of spermatogenesis, somatic imprints are erased and replaced with paternal imprinting (Hernandez et al., 2003). Reflecting these developmental changes, the testes-derived pluripotent cells, as well as EG cells, have erased somatic imprints and have partially established male imprints (Kanatsu-Shinohara et al., 2004). ES cells, on the other hand, maintain the somatic imprints found in the developing embryo and most normal adult tissues (Kanatsu-Shinohara et al., 2004; Tada et al., 2001). The significance of this difference is highlighted when one considers the phenotypes of ES cells, and the mice derived from them, which carry either no imprints (Holm et al., 2005) or dipaternal imprints (Hernandez et al., 2003). Transient deactivation of the DNA methytransferase Dnmt1 was used in one study to erase the imprints in mouse ES cells. Although these ES cells continued to self-renew in culture and contribute to high-grade chimeric mice, these chimeras developed widespread tumorigenesis within their first year (Holm et al., 2005). The case for dipaternal imprints is even more severe. Fibroblasts derived from androgenetic embryos (that is, produced from two male gametes) grow with an overtly transformed phenotype and ES cells derived from these embryos fail to contribute appreciably to chimeras (Hernandez et al., 2003). Although the maGSCs/MASCs are neither fully imprint-free nor entirely dipaternal, their intermediate state between these two extremes is still cause for concern. Hence, besides the obvious limitation in the exclusion of roughly half of the adult population, these questions and risks surrounding the imprinting status of testes-derived pluripotent cells makes them an inherently unattractive source of material for cell therapy.
5. Direct reprogramming
The newest addition to the reprogramming toolbox relies on the ability of a small set of defined transcription factors to directly push cells from one stable state to another on an “epigenetic landscape,” a concept first introduced by Conrad Hal Waddington (Slack, 2002; Waddington, 1957). Akin to the potential energy surfaces studied by physicists and chemists, Waddington's landscape consisted of hills and valleys representing various stable and unstable cellular phenotypes (see Figure 5A). Unlike a physical potential, however, each point on Waddington's surface represented a different gene expression pattern – that is, the epigenetic landscape exists in an N-dimensional space (where N is the number of genes encoded by the genome) with each axis representing levels of expression of a different gene. Thus, the potential represented by the height of each point on the surface is truly a cell fate potential, and, as in physics, the gradient at each point can be thought of a sort of force either pushing the cell towards a new fate (if on a hill) or maintaining it in a stable fate (if in a well). A pluripotent cell in a culture-induced self-renewing state, therefore, is like a marble sitting in a shallow well at the top of a hill, which, when allowed to differentiate will quickly roll down the hill towards one of many possible outcomes (see Figure 5A). Looking closer at the path a pluripotent cell takes towards a terminally differentiated state, such as a mature neuron, one could take a slice through this cellular potential surface and find that there may be many progenitor states of varying stabilities along the way (see Figure 5B).

(A) A Waddington-inspired schematic of the epigenetic landscape. Culture conditions will promote the self-renewal of a pluripotent cell, maintaining it in a shallow well at the top of a cellular potential hill. When allowed to differentiate, this cell will “roll” down the hill into one of many terminally-differentiated fates at lower potential. (B) A closer look at the path a pluripotent cell might take as it differentiates into a neuron, passing through a number of intermediate progenitor states of varying stability on the way. The line in (B) represents a slice through the surface shown in (A). (C) The process of direct reprogramming, like chemical catalyst, implicates a restructuring of the epigenetic landscape. Introduction of the transcription factor cocktail destabilizes the fibroblast identity while stabilizing the transition state. Because the retroviruses are shut down in the iPS cells, however, the potential of the pluripotent state remains unchanged.
While a generally downhill trend represents the normal process of development, the challenge of reprogramming is to move the cell either between stable wells or even to push it all the way back up the hill to a pluripotent state. The thought of using transcription factors to push cellular identity between stable states is hardly a new one, as Weintraub famously demonstrated in the 1980s that several cell types could be induced into a myoblast-like phenotype following the introduction of a single transcription factor, MyoD (Davis et al., 1987; Weintraub et al., 1989). A more recent study applying the same principle to directly reprogram B cells into macrophages reinforced the concept of direct reprogramming (Xie et al., 2004). Nevertheless, the widespread belief that the barrier was so high and the number of candidate factors (and possible combinations thereof) so great between terminally differentiated and pluripotent cell fates led to the assumption that, while direct reprogramming may work in principle, successful production of pluripotent stem cells in this fashion could not be achieved on any reasonable time-scale.
Despite this skepticism, an elegant 2006 study in mouse (Takahashi and Yamanaka, 2006), and its subsequent application to human (Takahashi et al., 2007; Yu et al., 2007), demonstrated that a straightforward, logical approach could be taken to the direct reprogramming of fibroblasts into what the authors called induced pluripotent-like stem (iPS) cells. Through a clever process of elimination, the authors found that iPS cells could be generated from both embryonic and adult fibroblasts after four transcription factors – Klf4, Sox2, Oct4, and cMyc – were introduced using murine retroviruses. Just as a chemical catylst changes the energy of a reaction's transition state, these virally-delivered transcription factors seem to reconstruct the cellular potential surface allowing for transitions to occur that were previously blocked by a barrier of prohibitive height (see Figure 5C). Because the retroviruses used by Yamanaka and colleagues are naturally silenced in ES (as well as iPS) cells, the resultant iPS cells must maintain their pluripotent state on their own and can therefore be said to have been truly reprogrammed (Maherali et al., 2007; Okita et al., 2007 Wernig et al., 2007), without artificially changing the potential of the pluripotent end-state of this process.
Although the first-generation iPS cells were not quite equivalent to ES cells in some regards (Rodolfa and Eggan, 2006; Takahashi and Yamanaka, 2006), technical refinements have since produced cells that are considerably more similar to normal ES cells in their transcriptional profiles, genome-wide epigenetic status, and even the ability to produce high-grade germline chimeras (Maherali et al., 2007; Okita et al., 2007; Wernig et al., 2007). Nevertheless, and unlike ntES cells, even these second-generation iPS cells are not entirely the same as fertilization-derived ES cells. For instance, reactivation of the retroviruses can lead to tumorigenesis in adult chimeras and their progeny (Nakagawa et al., 2008), highlighting a potentially serious limitation to their clinical utility. Additionally, it is concerning that there has been no demonstration to date of iPS cells giving rise to a mouse at term via tetraloid complementation, generally regarded as the most stringent test of pluripotency (Jaenisch and Young, 2008), although one midgestation embryo has been obtained (Wernig et al., 2007). The sources, and extent, of these differences between iPS and ES cells remain unclear, and determining how they can be overcome is a key goal of current efforts and a necessary prerequisite to applications in regenerative medicine.
The broad applicability of this approach has been shown in two mouse studies demonstrating that cells from the stomach and liver (Aoi et al., 2008), as well as mature B cells (Hanna et al., 2008), can also be reprogrammed into iPS cells. The latter study, in addition to directly demonstrating that iPS cells can be generated from terminally-differentiated cells, may have some medical relevance as peripheral blood is routinely isolated from patients. Nevertheless, skin punch biopsies are a relatively simple outpatient procedure, and banks of patient-derived fibroblast samples have already been established at several institutions. Indeed, recent results in our lab have shown that these patient cells can be used to generate patient-specific iPS cells and direct their differentiation into disease-relevant cell types (Dimos et al., 2008). While the generation of such cells holds great immediate promise for the in vitro modeling of degenerative diseases, particularly of those with little or no known genetic associations, the use of oncogenes and oncogenic retroviruses in their production precludes any therapeutic utility they might have. Although some researchers have made very preliminary progress towards eliminating the retroviruses by showing that iPS cells can be obtained without the oncogene cMyc (Nakagawa et al., 2008; Wernig et al., 2008) or replacing one or two transgenes with chemicals (Shi et al., 2008; albeit in a therapeutically irrelevant setting of neural progenitor cells), these studies represent only the first steps toward a more therapeutically-tractible system. And, despite an initial proof-of-principle cell therapy using iPS in a mouse sickle-cell anemia model (Hanna et al., 2007), directly reprogrammed cells won't be ready for clinical use until they can produced with either a transient introduction of genes, proteins, or small molecules, and any remaining differences between such next-generation iPS cells and bona fide ES cells are better understood.
6. Conclusions and future prospects
The decade following the cloning of Dolly has seen a revolution in the fields of stem cell biology and regenerative medicine. In that time, we have harnessed the ability to directly reshape the epigenetic landscape and impose a pluripotent phenotype upon a somatic genome. Each technique that has been devised to mediate this transition has provided new insights into the underlying process of nuclear reprogramming and brought us one step closer to the production of patient-specific pluripotent stem cells. However, each also poses considerable challenges that must be overcome before the dream of personalized regenerative medicine can be realized. The technical and logistical challenges of nuclear transfer are almost insurmountable for widespread application, as is the tetraploidy of hybrid ES cells generated upon stem-somatic cell fusion. Explantation of testes tissue to generate pluripotent stem cells may avoid such technical challenges, but suffers from an inherently limited patient-base and a questionable imprinting status. Direct reprogramming is at present the most promising avenue of research, but only time and further investigation will determine whether the retroviruses and oncogenes currently required can be eliminated and whether iPS cells can ever be fully equivalent to ES cells, allowing this new technique to replace NT as the gold standard of nuclear reprogramming.
Although there are challenges to overcome, the future for reprogramming seems quite promising. Patient-specific iPS cells generated thus far, even if not clinically applicable, can certainly be used for the modeling of disease phenotypes. This will for the first time allow researchers to investigate the onset and progression of degenerative diseases in cells that carry the entire complement of an affected individual's genes. Such modeling, of course, will be particularly powerful for studying the influence of environment on disease where no underlying genetic cause is known. And, while cells carrying retroviral integrations could never provide material for cell therapy, such in vitro disease models may still find clinical relevance as a substrate for drug discovery. Nor does it seem unreasonable to expect that retrovirus-free, clinically-relevant methods for producing iPS cells will be developed shortly, finally bringing about the first step on the road to regenerative medicine. With new technical advances will likely come a refined understanding of Waddington's epigenetic landscape and a greater capacity for more extensive manipulations of cell identity. Can Yamanaka's techniques be used to help guide the differentiation of pluripotent cells in culture towards a desired cell type? Or from one differentiated cell type into another, as some exciting recent results are beginning to indicate (Zhou et al., 2008)? Even with patient-specific pluripotent cells in hand, generation of the cell types relevant to their disease continues to be a major stumbling block in all but a handful of contexts, and these questions will undoubtedly play a dominant role in guiding future investigation.
Acknowledgements
K.T.R. is a graduate research fellow of the National Science Foundation and Sternlicht Fellow in Diabetes Research of the Harvard Stem Cell Institute.